Type 1 diabetes (T1D) is an autoimmune disease characterized by the destruction of insulin-producing pancreatic β-cells by their own immune system, resulting in lifelong insulin deficiency. Continuous exogenous insulin replacement therapy is the current standard of care for T1D. Transplantation of primary pancreatic islets or the entire pancreas is a viable remedy for managing patients with autoimmune T1D. However, this strategy is not feasible due to several obstacles, including a scarcity of donors, islet cells, and poor vascular engraftment of islets post-transplantation, as well as the need for prolonged immune suppression.
- type 1 diabetes
- autoimmune diseases
- pancreatic β-cells
- tolerance
- regulatory T cells
- immunotherapy
1. Introduction
2. Background of Autoimmune T1D
Autoimmune T1D occurs when our own immune system mistakenly destroys the insulin hormone-producing pancreatic β-cells in the Langerhans islets, schematically shown in Figure 1. T1D appears to have a genetic component and can be diagnosed in childhood and reported in later life, i.e., adulthood. Its causes are not completely understood or researched, and there is currently no likely treatment or cure. To survive, T1D patients must rely on injected or pumped insulin for the rest of their lives. T1D is found in both children and adults who exhibit symptoms such as frequent urination, dry mouth, increased thirst, itchy or dry skin, increased appetite, unexplained weight loss, and infections. Individuals with autoimmune T1D are typically diagnosed after exhibiting symptoms such as nausea, vomiting, extreme thirst, exhaustion, and/or malaise. As the body loses its ability to produce insulin, a hormone that allows the body to use the sugar found in everyday foods, known as glucose, as an energy source, patients with T1D must work closely with their endocrinologists to determine the insulin doses and lifestyle changes needed to manage their blood sugar levels. Autoimmune T1D patients are vulnerable to a variety of health issues, ranging from minor to severe; if you are not properly managed, your glucose levels will rise. Most T1D patients spend their lives managing it and have a problem with it, blood glucose levels are outside the clinically recommended beneficial range, which leads to potentially fatal hyperglycemia (high blood glucose) episodes and hypoglycemia (low blood glucose). Assume you do not maintain it and develop high blood sugar, which frequently leads to devastating health complications later in life, such as blindness, kidney failure, heart disease, and nerve damage, resulting in amputations.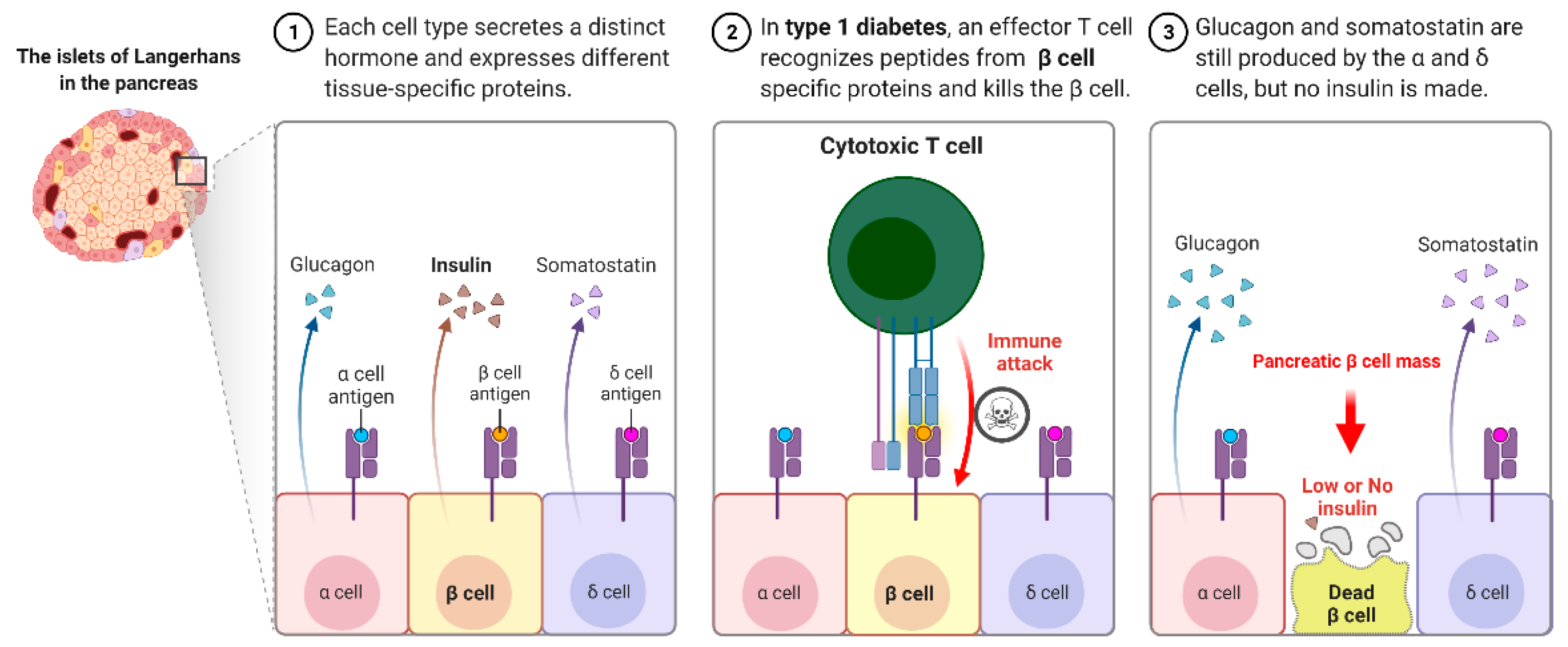
2.1. Immune Cells Involved in the Pathophysiology of Autoimmune Type 1 Diabetes
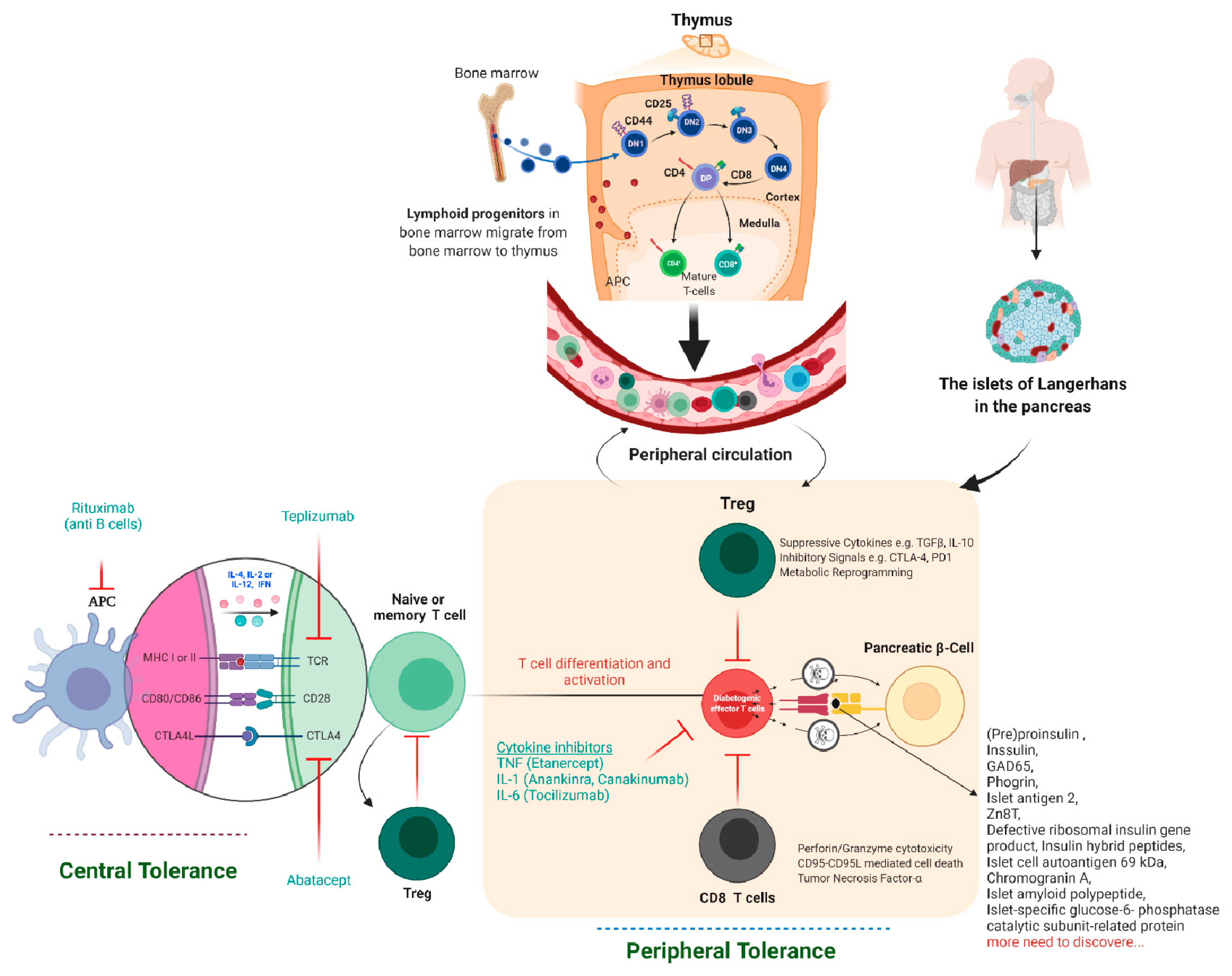
2.2. Immunotherapy-Based Approaches to Treating Autoimmune T1D
2.3. There Are a Few Obstacles to Immunotherapy-Based Treatment for Autoimmune T1D Cure
|
Therapeutic Agents |
Study/Authors and Intervention |
Outcome |
Citations |
|---|---|---|---|
|
T cell-based:
|
DEFEND-1, 2 (Otelexizumab) |
There was no EBV in the therapy group, but there was no statistically significant difference in 2-h MMTT AUC C-peptide at 12 months. |
[28] |
|
Protégé (Teplizumab) |
At 1 year, there was no significant difference in HbA1c1 < 6.5 percent or insulin dose < 0.5 U/kg per day: At year 2, AUC C-peptide in the high dose group was considerably greater than in the placebo group. |
||
|
AbATE (Teplizumab) |
The treatment group’s baseline adjusted AUC C-peptide reduced at year 2 was considerably lower. |
[31] |
|
|
B cell-based: The monoclonal anti-CD20 antibody, which blocks the B cell function |
Rituximab |
HbA1c lowers as the rate of C peptide declines and insulin levels decrease. |
|
|
Co-stimulation blockade |
TrialNet CTLA4-Ig (abatacept); CTLA-4-IgG1 chimeric protein acts as a decoy receptor for CD80/86 and blocks CD28-CD80/86 induced co-stimulation of T-cells |
Significantly higher stimulated C-peptide 2-h AUC in the treated group at the end of treatment and 1-year post-treatment |
|
|
TIDAL (alafacept); Alafacept: chimeric protein (2 LFA-3 molecule-IgG1) binds to CD2 and blocks T-cell-stimulation |
Significantly higher stimulated AUC C-peptide in the treatment group compared to placebo; insulin use lower in the treatment group |
[36] |
|
|
Cytokine-based: IL-2 agonist |
Aldesleukin; IL-2 maintains Treg population and function |
A dose-dependent elevation of Treg cells in the treatment group compared to placebo |
[37] |
|
TNF antagonism |
Etanercept |
HbA1c decreases while endogenous insulin production increases. |
[38] |
|
IL-1 receptor blockade |
Anakinra |
|
|
|
IL-1beta antagonism |
Canakinumab |
There was no C peptide reaction |
[39] |
|
IL-1 receptor blockade IL-1beta antagonism |
Anakinra/canakinumab |
Immunomodulation/reverse relationship between inflammation and C peptide stimulation |
[41] |
|
Anti-IL-6 therapy |
Tocilizumab in New-onset T1D (EXTEND) |
Ongoing study |
Clinical trial NCT02293837 |
|
Antigen-based therapy: |
Antigen-specific therapies may involve direct targeting of pathogenic T cells and/or boosting Tregs for bystander suppression |
Tregs were shown to be more prevalent in those who got a larger dose of oral insulin (62.5 mg) |
[42] |
|
Treg-based: |
Expansion of autologous Treg cells |
A subset of adoptively transferred Treg is still in circulation (25% of peak) at year 1, with no significant adverse effects. C-peptide preservation in those receiving a lower dose |
[20] |
|
DC-based: |
In T1D individuals who get their autologous DCs exhibited limited output. In this study, autologous DCs were given by infused via abdominal intradermal injections each 2 weeks apart |
The autologous DC-based therapy was very well tolerated; no important differences were seen in glycemia |
[37] |
|
Combination therapy |
|
C-peptide significantly increased at 30 months follow up; increased side effects |
[43] |
|
32% were insulin-free at 4 years, maintenance of C-peptide, but with increased side effects |
[44] |
||
|
Mean AUC C-peptide at 12 months was significantly higher in the study group compared to the placebo group |
Low-dose ATG + plus pegylated G-CSF [45] |
AbATE = Autoimmunity Blocking Antibody for Tolerance in Recently Diagnosed T1Ds, ATG = anti-thymocyte globulin, AUC = area under curve, CTLA-4 = cytotoxic T-lymphocyte-associated antigen, DEFEND = Durable Response Therapy Evaluation for Early or T1D = Type 1 Diabetes, EBV = Epstein Barr virus, G-CSF = granulocyte colony-stimulating factor, HbA1c = glycated haemoglobin, IL-2 = interleukin-2, LFA-3 = leukocyte function antigen-3, MMTT = mixed meal tolerance test, Pre-POINT = Primary intervention with Oral Insulin for Prevention of T1D in infants at high genetic risk, TCR = T-cell receptor, Teffs = T effector cells, TIDAL = Type 1 Diabetes with Alefacept, Tregs = T regulatory cells, DC = dendritic cells.
References
- Firestein, G.S.; Budd, R.C.; Harris, E.D.; McInnes, I.B.; Sergent, J.S. Kelley’s Textbook of Rheumatology, 8th ed.; WB Saunders: Philadephia, PA, USA, 2008.
- Goldman, L.; Ausiello, D.A. Cecil Medicine; Saunders Elsevier: Philadelphia, PA, USA, 2008.
- Herold, K.C.; Vignali, D.A.A.; Cooke, A.; Bluestone, J.A. Type 1 diabetes: Translating mechanistic observations into effective clinical outcomes. Nat. Rev. Immunol. 2013, 13, 243–256.
- Tisch, R.; McDevitt, H. Insulin-dependent diabetes mellitus. Cell 1996, 85, 291–297.
- Anderson, M.S.; Bluestone, J.A. THE NOD MOUSE: A Model of Immune Dysregulation. Annu. Rev. Immunol. 2005, 23, 447–485.
- Burn, P. Type 1 diabetes. Nat. Rev. Drug Discov. 2010, 9, 187–188.
- Głowińska-Olszewska, B.; Szabłowski, M.; Panas, P.; Żołądek, K.; Jamiołkowska-Sztabkowska, M.; Milewska, A.J.; Kadłubiska, A.; Polkowska, A.; Łuczyński, W.; Bossowski, A. Increasing Co-occurrence of Additional Autoimmune Disorders at Diabetes Type 1 Onset Among Children and Adolescents Diagnosed in Years 2010–2018—Single-Center Study. Front. Endocrinol. 2020, 11, 476.
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017, 8, 1898.
- Van Belle, T.L.; Coppieters, K.T.; Von Herrath, M.G. Type 1 diabetes: Etiology, immunology, and therapeutic strategies. Physiol. Rev. 2011, 91, 79–118.
- Atkinson, M.A. The Pathogenesis and Natural History of Type 1 Diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007641.
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016.
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986.
- Pepper, A.R.; Bruni, A.; Shapiro, A.J. Clinical islet transplantation: Is the future finally now? Curr. Opin. Organ Transplant. 2018, 23, 428–439.
- Bassi, R.; Fiorina, P. Impact of Islet Transplantation on Diabetes Complications and Quality of Life. Curr. Diabetes Rep. 2011, 11, 355–363.
- Pociot, F.; Lernmark, Å. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339.
- Harjutsalo, V.; Podar, T.; Tuomilehto, J. Cumulative Incidence of Type 1 Diabetes in 10,168 Siblings of Finnish Young-Onset Type 1 Diabetic Patients. Diabetes 2005, 54, 563–569.
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for Islet Autoimmunity among Monozygotic Twins. N. Engl. J. Med. 2008, 359, 2849–2850.
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707.
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343.
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189.
- Ali, M.A.; Liu, Y.-F.; Arif, S.; Tatovic, D.; Shariff, H.; Gibson, V.B.; Yusuf, N.; Baptista, R.; Eichmann, M.; Petrov, N.; et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 2017, 9, eaaf7779.
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 2016, 530, 434–440.
- Skyler, J.S.; Pugliese, A. Immunotherapy Trials for Type 1 Diabetes: The Contribution of George Eisenbarth. Diabetes Technol. Ther. 2013, 15, S2-13–S2-30.
- Feutren, G.; Assan, R.; Karsenty, G.; Du Rostu, H.; Sirmai, J.; Papoz, L.; Vialettes, B.; Vexiau, P.; Rodier, M.; Lallemand, A.; et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset: Results of a multicentre double-blind trial. Lancet 1986, 328, 119–124.
- Sigal, N.H.; Dumont, F.J.; Cyclosporin, A. FK-506, and rapamycin: Pharmacologic probes of lymphocyte signal transduction. Annu. Rev. Immunol. 1992, 10, 519–560.
- Canadian-European Randomized Control Trial Group. Cyclosporin-induced remission of IDDM after early intervention: Association of 1 yr of cyclosporin treatment with enhanced insulin secretion. Diabetes 1988, 37, 1574–1582.
- Füchtenbusch, M.; Kredel, K.; Bonifacio, E.; Schnell, O.; Ziegler, A.G. Exposure to exogenous insulin promotes IgG1 and the T-helper 2-associated IgG4 responses to insulin but not to other islet autoantigens. Diabetes 2000, 49, 918–925.
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural basis of assembly of the human T cell receptor–CD3 complex. Nature 2019, 573, 546–552.
- Long, S.A.; Thorpe, J.; DeBerg, H.A.; Gersuk, V.; Eddy, J.A.; Harris, K.M.; Ehlers, M.; Herold, K.C.; Nepom, G.T.; Linsley, P.S. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci. Immunol. 2016, 1, eaai7793.
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613.
- Dolgin, E. Anti-CD3 drug keeps diabetes at bay. Nat. Biotechnol. 2019, 37, 1099–1101.
- Wang, X.; Ni, L.; Chang, D.; Lu, H.; Jiang, Y.; Kim, B.-S.; Wang, A.; Liu, X.; Zhong, B.; Yang, X.; et al. Cyclic AMP-Responsive Element-Binding Protein (CREB) is Critical in Autoimmunity by Promoting Th17 but Inhibiting Treg Cell Differentiation. EBioMedicine 2017, 25, 165–174.
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-Hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68.
- Wang, M.; Yang, L.; Sheng, X.; Chen, W.; Tang, H.; Sheng, H.; Xi, B.; Zang, Y.Q. T-cell vaccination leads to suppression of intrapancreatic Th17 cells through Stat3-mediated RORγt inhibition in autoimmune diabetes. Cell Res. 2011, 21, 1358–1369.
- Stifter, K.; Schuster, C.; Schlosser, M.; Boehm, B.O.; Schirmbeck, R. Exploring the induction of preproinsulin-specific Foxp3+ CD4+ Treg cells that inhibit CD8+ T cell-mediated autoimmune diabetes by DNA vaccination. Sci. Rep. 2016, 6, 29419.
- Klein, L.; Robey, E.A.; Hsieh, C.-S. Central CD4+ T cell tolerance: Deletion versus regulatory T cell differentiation. Nat. Rev. Immunol. 2019, 19, 7–18.
- Kitashima, D.Y.; Kobayashi, T.; Woodring, T.; Idouchi, K.; Döbel, T.; Voisin, B.; Adachi, T.; Ouchi, T.; Takahashi, H.; Nishifuji, K.; et al. Langerhans Cells Prevent Autoimmunity via Expansion of Keratinocyte Antigen-Specific Regulatory T Cells. EBioMedicine 2018, 27, 293–303.
- Boardman, D.; Levings, M.K. Cancer immunotherapies repurposed for use in autoimmunity. Nat. Biomed. Eng. 2019, 3, 259–263.
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62.
- Tack, C.J.; Kleijwegt, F.S.; Van Riel, P.L.C.M.; Roep, B.O. Development of type 1 diabetes in a patient treated with anti-TNF-α therapy for active rheumatoid arthritis. Diabetologia 2009, 52, 1442–1444.
- Mastrandrea, L.; Yu, J.; Behrens, T.; Buchlis, J.; Albini, C.; Fourtner, S.; Quattrin, T. Etanercept Treatment in Children With New-Onset Type 1 Diabetes: Pilot Randomized, Placebo-Controlled, Double-Blind Study: Response to Peters. Diabetes Care 2009, 32, e154.
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15.
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-α therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746.
- Faustman, D.L. TNF, TNF inducers, and TNFR2 agonists: A new path to type 1 diabetes treatment. Diabetes/Metab. Res. Rev. 2018, 34, e2941.
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.A.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328.