Apoptosis is an evolutionarily conserved and tightly regulated cell death pathway. Physiological cell death is important for maintaining homeostasis and optimal biological conditions by continuous elimination of undesired or superfluous cells. The BH3-only pro-apoptotic members are strong inducers of apoptosis. The pro-apoptotic BH3-only protein Noxa activates multiple death pathways by inhibiting the anti-apoptotic B-cell lymphoma-2 (Bcl-2) l-2 family protein, Mcl-1, and other protein members leading to Bax and Bak activation and mitochondrial outer membrane permeabilization (MOMP) MOMP. On the other hand, Puma is induced by p53-dependent and p53-independent apoptotic stimuli in several cancer cell lines. Moreover, this protein is involved in several physiological and pathological processes, such as immunity, cancer, and neurodegenerative diseases. Future heat shock research could disclose the effect of hyperthermia on both Noxa and BH3-only proteins. This suggests post-transcriptional mechanisms controlling the translation of both Puma and Noxa mRNA in heat-shocked cells.
- Noxa
- Puma
- Bcl-2 proteins
- p53
- heat-shock
- caspase
- ion channels
- apoptosis
1. Introduction
2. Bcl-2 Proteins and the Regulation of Mitochondrial Outer Membrane Permeability
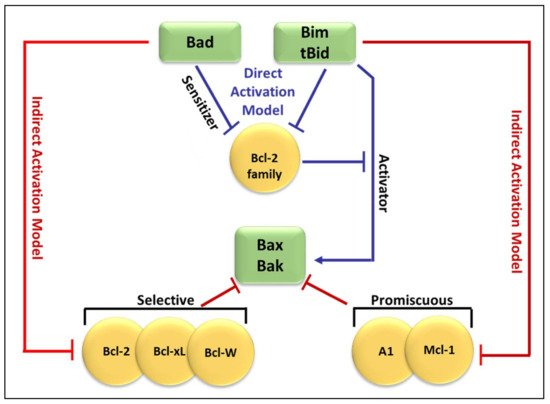
Molecular insights into apoptosis first emerged during the 1980s and 1990s from a convergence of mammalian cancer cytogenetics and genetic studies on developmentally programmed cell death in Caenorhabditis elegans [35][38]. The previously unknown gene Bcl-2 was identified from the breakpoint region of a recurrent chromosomal translocation (18q21) in human follicular lymphoma [7][36][7,39]. The Bcl-2 family of proteins are central regulators of stress-induced apoptosis as they control diverse survival and death signals that are generated inside and outside the cell [37][38][40,41]. Bcl-2 proteins are found in all cells and consist of 12 distinct members, many of which are expressed as alternate isoforms. Structurally, they all contain at least one of the conserved sequence motifs called Bcl-2 BH domains. This family is functionally subdivided into two classes based on their activity and number of BH domains: anti-apoptotic and pro-apoptotic proteins including the BH3-only members [16][38][39][19,41,42]. The mutual interaction between pro-apoptotic and anti-apoptotic members establishes the threshold that determines whether a cell should survive or die [40][43]. In a sense, they act as checkpoints through which survival and death signals must pass to elicit the cell’s fate as they control stress-induced cell death [41][44]. The anti-apoptotic Bcl-2 members, including Bcl-2 itself, Bcl-xL, Mcl-1, Bcl-W, and A1/BFL-1 share four conserved BH domains of structural homology. These proteins prevent cell death against diverse cytotoxic signals, both physiological and imposed. This maintains the integrity of the ER, mitochondrial, and nuclear membranes, thus protecting cells from apoptosis [42][43][45,46]. The proximity of BH 1, 2, and 3 form a hydrophobic pocket that operates as a receptor for the BH3 domain of the pro-apoptotic BH3-only members. The anti-apoptotic Bcl-2 proteins work together to inhibit the release of cytochrome c from the mitochondria by preventing the activation of the pro-apoptotic members Bax and Bak in the OMM [41][44]. The anti-apoptotic proteins’ normal function is to prevent inappropriate cell death and therefore requires the neutralization of the pro-apoptotic proteins by the anti-apoptotic members [44][47]. Just as the anti-apoptotic Bcl-2 proteins promote tumourigenesis when deregulated, the pro-apoptotic members function as tumor suppressors. The pro-apoptotic Bcl-2 members are divided into multi-domain effector proteins, such as Bax, Bak, and the less well-known Bok, as well as the large subgroup of BH3-only proteins, all of which trigger or sensitize the cell to apoptosis [45][46][47][48,49,50]. The pro-apoptotic effector proteins Bax, Bak, and Bok possess three BH domains and adopt similar globular structures: a helical bundle surrounding a central hydrophobic core helix [41][48][49][44,51,52]. This groove constitutes a crucial surface for interactions with the BH3 domain of pro-apoptotic members of the Bcl-2 family [35][38]. These interactions primarily occur on intracellular membranes, such as that of the OMM, where many of the Bcl-2 family members are directed by their carboxy-terminal hydrophobic TM domain [50][53]. Unlike the anti-apoptotic members, the active conformation of Bax/Bak damages rather than protects the OMM of stressed cells. This yields the formation of pores leading to membrane dysfunction [51][54]. The most studied pro-apoptotic proteins Bax and Bak exist in an inactive monomeric state in healthy cells. In stressed cells, they are major inducers of mitochondrial outer membrane permeabilization (MOMP) [52][55]. Recent studies have focused on the effects of the Bcl-2 family members on MOMP. Although over a decade has passed since the discovery of the BH3-only protein Puma, the question of how this protein activates Bax/Bak, consequently leading to MOMP, remains unresolved. At present, two mechanisms have been proposed concerning the relationship of the Bcl-2 protein family members employing direct or indirect activation of the mitochondrial apoptotic pathway, shown in Figure 12 [41][44][51][44,47,54]. Two models are explaining Bak/Bax activation through the interaction of the Bcl-2 protein family members: the direct activation model and the indirect activation model.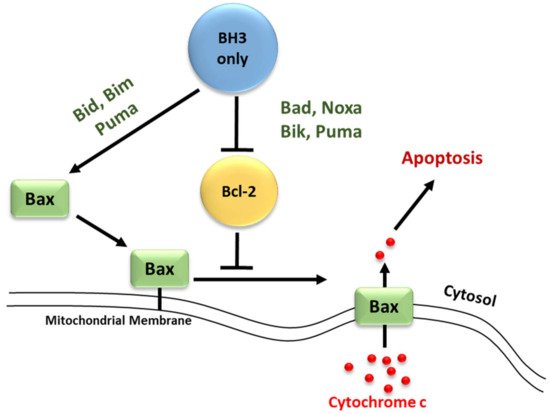
3. Regulation of Apoptosis
3.1. Regulators of Cell Death: Caspases
Caspases (Cysteinyl aspartate-specific proteases) are a family of proteins activated by a variety of stimuli representing a vital step for the induction of apoptosis. These proteases initiate and control the cellular death pathway by cleaving a diverse set of cellular proteins [57][68].
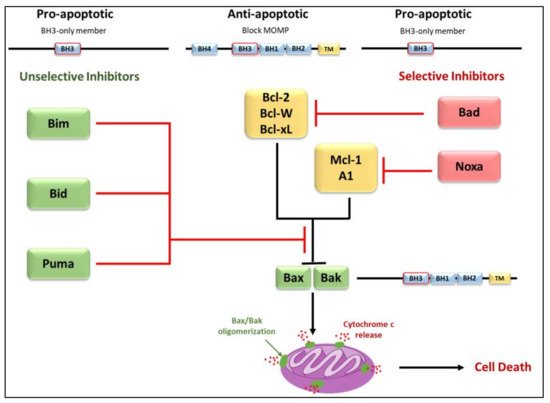
3.2. Regulators of Cell Death: Bcl-2 Family Proteins
3.3. Elucidation of the Caspases Reaction Mechanism
Caspases stand for Cysteine (Cys)-dependent aspartate-directed proteases are a sub-branch of the enzymes family proteases which possess an indispensable role in apoptosis. The commonly proposed mechanism is divided into two principal parts. It describes the cleavage of the peptide bond (amide function).
-
Once, again the alkaline property of one of the nitrogen atoms in the imidazolic part of His deprotonates a water molecule.
-
This deprotonation contributes to the formation of a hydroxide. The strong alkaline and nucleophilic property of the hydroxide contributes to the attack of the electrophilic site of the carbonyl function. This yields a second tetrahedral intermediate.
-
α-thio protonation: Similarly, to the third step of phase 1, the sulfur in I2 constitutes a good leaving group. This enhances the possibility of protonation of the α-thio
-
Formation of the carboxylic acid by regeneration of His and Cys counterparts.
-
The already formed hydrogen bond between His and the first water molecule will favor the deprotonation of the latter. The yielded hydroxide attacks the acyl-enzyme complex on the carbonyl moiety.
-
The yielded alkoxide in I3 will attack the proton already captured by the His part in (a). This will form a germinal diol (I4; Figure 46).
- moiety by the previously protonated nitrogen of the His residue.
- The carboxylate function of the side-chain aspartate will attack one of the hydrogens of the diol. The thiol, acting as a good leaving group, will enhance the possibility of the formation of a carbonyl bond; thereby a carboxylic acid and a thiolate (I5; Figure 46). The computational investigation shows that for the attack of the water molecule, a free energy barrier of about 19 ± 4 kcal/mol must be overcome, these trends are following the experimental results of Sulpizi et al. [64][88].
- Nucleophilic activation: the alkaline property of one of the nitrogen atoms, in the imidazolic part of Histidine (His), deprotonates the hydrogen in the thiol (-SH) group of the Cys residue, yielding a thiolate.
-
Thiolate nucleophilic attack on carbonyl: the carbonyl group of the aspartic peptide bond undergoes a nucleophilic attack by the yielded thiolate in the latter step. This contributes to the formation of a first tetrahedral intermediate.
-
α-amino protonation: the amine group of I1 constitutes a good leaving group. This will enhance the possibility of the protonation of the α-amino moiety by the previous protonated nitrogen of the His residue.
-
Formation of the covalent adduct: the acyl-enzyme complex and the cleavage of the peptide bond.