Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xuehui Fan and Version 2 by Vivi Li.
Takotsubo syndrome (TTS) is identified as an acute severe ventricular systolic dysfunction, which is usually characterized by reversible and transient akinesia of walls of the ventricle in the absence of a significant obstructive coronary artery disease (CAD). Patients present with chest pain, ST-segment elevation or ischemia signs on ECG and increased troponin, similar to myocardial infarction. Currently, the known mechanisms associated with the development of TTS include elevated levels of circulating plasma catecholamines and their metabolites, coronary microvascular dysfunction, sympathetic hyperexcitability, inflammation, estrogen deficiency, spasm of the epicardial coronary vessels, genetic predisposition and thyroidal dysfunction.
- Takotsubo syndrome
- pathophysiological mechanism
- human-induced pluripotent stem cell-derived cardiomyocytes
- catecholamines
- precision medicine
1. Introduction
Takotsubo syndrome (TTS), also known as Takotsubo cardiomyopathy (TTC) or broken heart syndrome or stress-induced cardiomyopathy, is a type of acute heart failure syndrome characterized by an acute, transient and reversible left ventricular (LV) systolic dysfunction with apical ballooning and ST-segment elevation or T-wave inversion in the absence of obstructive coronary artery disease (CAD), which is often associated with emotional or physical stress [1][2][1,2]. TTS has been reported in adults of all ages and even in children [3]. TTS was first described in Japan in 1990, in five cases of typical cardiac arrest patients suffering from chest pain with abnormal electrocardiogram, similar to acute myocardial infarction (AMI), but without evidence of coronary artery stenosis on angiography (CAG) [4]. Importantly, the major criteria to differentiate TTS from AMI are the absence of significant obstructive CAD contributing to the extent of contraction abnormalities [5][6][5,6], and the recovery of the wall motion abnormality and ejection fraction abnormality. TTS and neurogenic stunning (NS) share many common features, although there are differences in the clinical and laboratory features between them [7][8][7,8]. For example, apical hypokinesia is more common in Takotsubo, whereas basal hypokinesia is typical of patients with NS [9]. The development of neurogenic cardiomyopathy (NC) is mainly associated with an elevated plasma norepinephrine (NE) level, which is mediated by neuronal NE rather than adrenal epinephrine (EPI) [10]. Both a surge in catecholamine secretion in the first hour of brain death and a deficiency in the following hours are causes of cardiac stunning and altered vascular reactivity in NS [11].
The suggested mechanisms associated with the development of TTS include elevated levels of circulating plasma catecholamines and its metabolites, coronary microvascular dysfunction, sympathetic hyperexcitability, inflammation, estrogen deficiency, basal hypercontractility with left ventricular outflow tract obstruction and signal trafficking/biased agonism, spasm of the epicardial coronary vessels, genetic predisposition and thyroidal dysfunction [12][13][14][15][16][17][12,13,14,15,16,17]. Since the pathogenesis of TTS contains probably a combination of multiple factors, the precise pathogenesis of TTS is difficult to fully explore. Contemporary analyses show that 20–25% of TTS patients have received beta-blocker drugs during their initial event and 40% during a second episode. However, no drug treatment has been found to be effective to prevent the recurrence or improve the outcome of TTS [18]. The lack of optimal treatment for TTS leads to the necessity of the application of experimental models or platforms for studying TTS. Several models, mainly focusing on animal models and human-induced pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) models, have been used to simulate the disease and investigate possible pathogenic mechanisms. Each model has its own advantages and disadvantages.
2. Experimental Models of TTS
2.1. Animal Models and Mechanistic Studies
Experimental animal models may be a valuable tool for studying human diseases, which can be potentially used to clarify one or more specific questions. Compared with human models, animal models are relatively easier to manage and highly available. In addition, many animals are suitable for studying human diseases due to their high similarity in terms of anatomical basis and physiological functions with humans. Thus, the use of animal models can undoubtedly provide new insights for studying and elucidating the important pathophysiological mechanisms of TTS.
In recent years, some researchers have already used animal models treated with high doses of catecholamines or adrenoceptor agonists or immobilization (IMO) to stimulate some or all of the abovementioned clinical manifestations of TTS. IMO in rats was used to study psychological stress, which is a successful model of TTS in rats [19][20][21][22][23][24][25][26][27][152,153,154,182,183,184,185,186,187]. IMO in rats triggered a transient and reversible reduction of LV contraction, including LV apical ballooning prevented by pretreatment with an adrenoceptor blocker, suggesting that the activation of β1 adrenergic receptors in the heart and the activation of α1 adrenergic receptors in the aorta are the primary cause of TTS [23][28][29][183,188,189].
Furthermore, ovariectomized (OVX) and estradiol-supplemented ovariectomized female rats (OVX+E) with IMO stress were used as an animal model of TTS, finding that the percentage contraction in the left ventriculography of OVX rats was significantly reduced in response to stress, while the mRNA expression of c-fos, a marker of cellular activation, in the OVX+E rats was significantly decreased in the paraventricular hypothalamic nucleus, adrenal gland and LV, suggesting that an increase of estrogen can attenuate the emotional stress-induced hypothalamo–sympatho–adrenal outflow from the central nervous system to the target organs [21][154].
Of note, the isoprenaline (ISO)-induced female TTS rat model needs a higher triggering dose and has a lower mortality than that of male TTS rats [30][190]. This may be related to the fact that estrogen has a vasodilator effect through the induction and activation of endothelial nitric oxide synthase, and the reduction of estrogen may interfere with coronary microcirculation by indirect action on the nervous system and by direct action on the heart vessels [31][27][119,187].
Another possible mechanism is that estrogen protects the myocardium from TTS by increasing the activity of the β2 AR-Gas signaling pathway and reducing the concentration of catecholamines in plasma [32][191]. Repeated intravenous infusion with epinephrine in cynomolgus monkeys induced LV dysfunction with apical ballooning and wall motion abnormalities, which mimics that of TTS in clinical cases [33][192]. This model is of great value in understanding the pathogenesis of TTS associated with sympathomimetic hyperfunction in non-human primate models. In addition, a worthy and novel experimental model for inverted TTS-like cardiomyopathy is the ventricular tachyarrhythmia-related inverted cardiomyopathy in rabbits with vagal stimulation. Cardiac lesions related to ventricular tachyarrhythmias were involved in the basal portion, mitral valve and papillary muscles but not the apex [34][193]. Nevertheless, an α-adrenergic blockade reduced the development of the adrenaline-induced cardiac basal lesion but did not affect the structural and functional alterations [35][194]. Importantly, epinephrine-specific β2AR-G(i) signaling may have evolved as a cardioprotective strategy to limit catecholamine-induced myocardial toxicity during acute stress, while α2AR/Gi-dependent signaling attenuates myosin-binding protein-C (MyBP-C) phosphorylation and contractility in the anterior wall (AW) through an epinephrine surge in TTS rats [16][22][16,182]. However, no statistically significant difference of the β2AR-dependent cAMP levels was observed between the apical and basal cells [32][191].
Moreover, a single injection of isoproterenol in mice could induce TTS-like regional dyskinesia, and the study demonstrated that lipotoxicity is closely related to catecholamine-induced myocardial dysfunction, including neurogenic stunning, metabolic stunning and electrophysiological stunning [36][195]. The direct inhibition of myocardial ApoB lipoproteins and subsequent reduction in lipid export by the supraphysiological level of catecholamines may result in cardiac lipotoxicity.
Emotional stress and a surge of catecholamine upregulated oxidative stress-related factor-heme oxygenase-1 (HO-1) in the heart, while the blockade of α- and β-adrenoceptors attenuated this effect [19][152], suggest that oxidative stress plays an important role in the occurrence and development of TTS. Recent evidence indicated that TTS patients suffered from acute endogenous catecholamine release, which may trigger oxidative stress and inflammation. The apoptosis of inflammatory cells was visible in the whole course of TTS, while the apoptosis of endothelial cells appeared only in the recovery phase [37][196]. ISO significantly increased the levels of reactive oxygen species (ROS) in the setting of TTS, leading to the injury of myocardial cells, mitochondrial dysfunction, acute Ca2+ overload, TLR4/NF-κB signaling alterations, dysregulation of the glucose and lipid metabolic pathways represented by decreases in the final glycolytic and β-oxidation metabolites and reduced availability of Krebs cycle intermediates. It was shown that icariin, an antioxidant and anti-inflammatory agent, prevented ISO-induced TTS-like cardiac dysfunction in rats by suppressing the TLR4/NF-κB pathway and long-term inhibition of PI3K/AKT/mTOR expression and by reducing the mitochondrial ROS and oxidative stress-induced apoptosis, which provides new insights into the protective effect against myocardial dysfunction in TTS rats [38][39][40][41][42][197,198,199,200,201].
Early treatment with isoflurane reduced left ventricular dyskinesia and improved the survival rate of experimental TTS, while H2S reduced ROS formation by reducing NADPH oxidase [43][44][202,203]. GPER, azelnidipine, Tempol and amlodipine also played a protective role for TTS [20][26][45][46][153,186,204,205]. In terms of mechanism, this effect is mediated by balancing the coupling of β2AR with the Gαs and Gαi signaling pathways [45][204].
In the TTS animal models, the subjects must be killed for testing certain biomarkers. Interestingly, speckle tracking echocardiography (STE) and global longitudinal strain (GLS) can be used to quantitatively detect subtle myocardial abnormalities and provide a reliable, noninvasive method to predict early myocardial injury in stress cardiomyopathy (SCM) rats, showing that ISO can reduce GLS and circumferential (GCS) strains of males and females [25][47][185,206].
The animal models of TTS are mainly based on monkeys, rabbits, rats and mice, which can be used to simulate TTS to a certain extent through immobilization, repeated intravenous infusion of an epinephrine overdose, vagal stimulation, intraperitoneally isoprenaline and other methods [19][20][21][22][23][24][25][26][27][28][30][32][33][34][35][36][38][39][40][41][42][43][44][45][46][47][152,153,154,182,183,184,185,186,187,188,190,191,192,193,194,195,197,198,199,200,201,202,203,204,205,206]. Many live-stranded cetaceans and terrestrial wildlife experienced capture myopathy, which was similar to TTS [48][49][207,208]. Nevertheless, the animal models of TTS cannot fully replicate the characteristics of human TTS and clarify the mysterious etiology of its human manifestations. There is a huge species difference between them and humans. In addition, different aspects were not explored in these animal models, including the corresponding mild-to-moderate release of cardiac biomarkers, the natural course of the electrocardiographic manifestations, the complete recovery of LV function and the presence and the degree of myocardial edema, all of which have been observed in patients with TTS. Therefore, other models are essential, although these animal models can simulate TTS to a certain extent and provide the basis for pathophysiological studies.
2.2. Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs) Models
The hiPSC-CM possess a lot of advantages in modeling genetic cardiac disorders like long-QT syndrome, short QT syndrome, arrhythmogenic right ventricular cardiomyopathy (ARVC), Brugada syndrome and familial hypertrophic cardiomyopathy (HCM) [50][51][52][53][54][55][56][57][58][59][60][209,210,211,212,213,214,215,216,217,218,219]. First, hiPSC-CMs possess a human gene background, avoiding possible effects resulting from the gene differences between humans and animals. Second, hiPSC-CMs provide a better tool for cardiac drug screening and ion channel research compared with Xenopus oocytes, human embryonic kidney (HEK) cells and Chinese hamster ovary (CHO) cells, which lack cardiac ion channel macromolecular complexes. Third, disease-specific or person-specific hiPSC-CMs can be generated, and patient-specific mechanistic study and drug testing can be performed, suggesting their significance for precision medicine. Additionally, genome editing combined with patient-specific iPSC-CMs has allowed exploring the genotype–phenotype correlation of unknown gene mutations or variants in patients.
So far, only several TTS-relating studies using hiPSC-CMs have been reported [61][62][45][25,95,204]. In the last few years, TTS was not regarded as a genetic disease. However, increasing evidence suggests the involvement of genetic factors in TTS. For example, highly relevant variants, such as RBM20, encoding RNA-binding motif protein 20, or CASQ2, encoding calsequestrin 2, exist in TTS patients [63][132]. Furthermore, acquired long QT syndrome has been described in up to 70% of TTS cases and is associated with life-threatening arrhythmias (LTA) triggered by concomitant atrial fibrillation. El-Battrawy et al. used hiPSC-CMs to investigate the toxic effects of catecholamine on cellular electrophysiological properties and the protective effects of estrogen, finding that estrogen may reduce the sensitivity of cardiomyocytes to catecholamine by reducing adrenoceptor expression [14]. The pathogenesis process of arrhythmias was induced by catecholamine excess, which caused the overstimulation of adrenoceptors, increase in ROS production, ion channel dysfunction (enhancement of late INa), APD/QT prolongation and tachyarrhythmias. Their study suggests that ROS-blocker, β-blocker and late sodium channel blocker may be potential alternatives to treat arrhythmias induced by catecholamine excess.
Another TTS model in hiPSC-CMs was developed with high doses of epinephrine, identifying that G1/estrogen (E2) alleviated epinephrine-induced cardiac damage via reducing the brain natriuretic peptide in plasma and released lactate dehydrogenase into the culture supernatant [45][204]. Huang et al. used a toxic concentration of epinephrine to mimic the setting of TTS, finding that high concentrations of epinephrine prolonged APD and induced arrhythmia events [62][95]. The effect of epinephrine was attenuated by alpha-adrenergic receptor blockers phentolamine [62][95]. An activator of alpha-1, but not alpha-2, receptor mimicked epinephrine effects, indicating that the alpha-1 receptor contributes to arrhythmogenesis of TTS. Further, it was revealed that NADPH-ROS-PKC signaling mediated the effects of the alpha-1 receptor [62][64][95,220], which may provide new insights for the treatment of TTS.
Of note, whether hiPSC-CMs from TTS patients can recapitulate some features of the disease need to be investigated. Borchert et al. used, for the first time, hiPSC-CMs generated from two TTS patients for the study, revealing that β-adrenergic signaling, including the cAMP response and cAMP-dependent PKA activity, was increased in TTS-iPSC-CMs after treatment with high levels of catecholamines [63][132]. The enhanced β-adrenergic signaling in TTS-iPSC-CMs under catecholamine-induced stress increased the cardiac stress marker NR4A1 expression [63][132]. These data strongly support applications of hiPSC-CMs for TTS studies and also suggests the possible involvement of genetic factors for the pathogenesis of TTS. Catecholamine-treated hiPSC-CMs or TTS-specific iPSC-CMs mimicked features consistent with those found in individuals with TTS. Additionally, TTS-iPSC-CMs provide a promising and reliable cell source for elucidation of the pathophysiological mechanism, drug screening, ion channel research and gene function research (Figure 12).
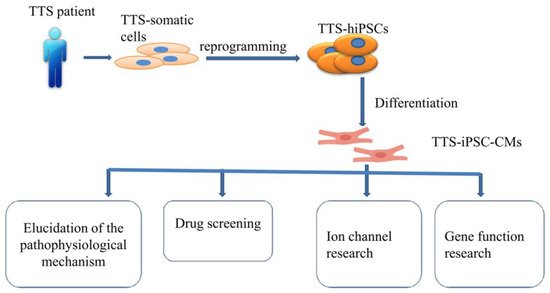
Figure 12.
Overview of human-induced pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) model.
However, hiPSC-CMs have several limitations, such as a lack of t-tubular network, polygonal shapes and rhythmic automaticity. Importantly, to overcome the limitations of a relatively immature fetal-like phenotype of hiPSC-CMs, extending the cell culture time [65][221], using a medium containing galactose and fatty acids [66][222], electrical field stimulation [67][223], electric pacing and mechanical stimulation [68][224], extracellular matrix [69][225] and 3D cardiac tissue with electric stimulation [70][226] have been used to improve the maturity of hiPSC-CMs.
HiPSC-CMs can overcome the limitations of artificial animal or other in vitro TTS models. Therefore, hiPSC-CMs provide a very helpful and easily available disease research model for translational medicine, which can bridge the gap between basic research and clinical disease.
2.3. Other Models
In addition to hiPSC-CMs, H9C2 cells can also be used to study the pathophysiological mechanism of TTS. Pretreatment with Tempol can reduce the production of reactive oxygen species and the deposition of lipid droplets and protect the mitochondrial function by reducing mitochondrial swelling, which provides a theoretical basis for Tempol to prevent isoproterenol-induced TTS [46][205]. Additionally, a computational model of the rat LV in TTS was used to investigate the mechanisms of the typical shape of the ventricle observed in TTS, finding that three potential dominant mechanisms are related to the effects of β-adrenergic stimulation, for example, the apical–basal variation of calcium transients, apical–basal variation of calcium sensitivity and apical–basal variation in maximal active tension [71][227]. The data from computational models can be used to interpret future experimental data and provide a theoretical basis for basic experiments.
At present, from the study of TTS models, scientists have demonstrated that some known mechanisms related to the pathogenesis of TTS from those models include a surge in catecholamines, endothelial dysfunction, estradiol levels, coronary microvascular dysfunction, apoptosis in CMs, etc. However, the animal or cell models of TTS that can mimic the characteristics of clinical TTS are limited. Importantly, researchers have made a certain level of discoveries from animal models to cellular models, which provides strong evidence for elucidating the pathophysiological mechanism of TTS (Table 1).
Table 1.
Experimental models for TTS.
| Model | Method | Main Finding | |
|---|---|---|---|
| Animal model | Rats [20][22][23][26][27][28][29][153,182,183,186,187,188,189] | Immobilization (IMO) | (1) The activation of β1 adrenergic receptors in the heart and the activation of α1 adrenergic receptors in the aorta were the primary cause of TTS (2) The reduction of estrogen may interfere with coronary microcirculation and may be involved in the primary cause of TTS by indirect action on the nervous system and by direct action on the heart (3) α2AR/Gi-dependent signaling attenuated myosin-binding protein-C(MyBP-C) phosphorylation and contractility in the anterior wall (AW) through an epinephrine surge in TTS rats |
| Ovariectomized (OVX) and estradiol-supplemented ovariectomized female rats [19][21][152,154] | Immobilization (IMO) | (1) The reduction of LV contractility and the increase of heart rate in response to emotional stress were attenuated by supplement of estradiol in the ovariectomized rats. (2) Emotional stress and a surge of catecholamine upregulated heme oxygenase-1 (HO-1) |
|
| Cynomolgus monkeys [33][192] | Intravenous infusion of epinephrine overdose | LV dysfunction with apical ballooning and wall motion abnormalities | |
| Rabbits [34][193] | Vagal stimulation | The cardiac lesions related to ventricular arrhythmias were involved in the basal portion, mitral valve, and papillary muscles but not the apex | |
| Mice [25][36][47][185,195,206] | A single dose injection of isoprenaline | (1) Lipotoxicity was closely related to catecholamine-induced myocardial dysfunction, including neurogenic stunning, metabolic stunning, and electrophysiological stunning (2) ISO reduced GLS and circumferential (GCS) strains of males and females |
|
| Rats [30][35][38]46][190,194[,19739],198[40],199[41],200[42],201[43],202[44],203[,205] | Isoprenaline (ISO) | (1) TTS rats had significantly lower left ventricular end-diastolic pressure and significantly better estimates of cardiac function. (2) Its apical perfusion was not impaired in the early stage of TTS (3) ISO significantly increased the levels of reactive oxygen species (ROS) in the setting of TTS (4) Early treatment with isoflurane could reduce LV dyskinesia and improve the survival rate of experimental TTS (5) GPER, azelnidipine, Tempol and amlodipine also played a protective role for TTS |
|
| Rats [45][204] | Epinephrine | GPER played a protective role against TTS | |
| hiPSC-CMs models | hiPSC-CMs models [14] | Isoprenaline | Estradiol had protective effects against catecholamine excess and hence reduction in estrogen level may increase the risk of acquired long QT syndrome in TTC |
| hiPSC-CMs models [45][204] | Epinephrine | Knockdown of GPER by siRNA abolished E2 effects on increasing ICa-L and action potential duration in the stress state | |
| hiPSC-CMs models [62][95] | Epinephrine | High concentrations of epinephrine inhibited the depolarization rate in hiPSC-CMs, the duration of action potentials and induced arrhythmia events while the effect of epinephrine was attenuated by alpha-adrenergic receptor blockers-phentolamine | |
| TTS-iPSC-CMs [63][132] | The β-adrenergic signaling, including cAMP response and cAMP-dependent PKA activity, was increased in TTS-iPSC-CMs | ||
| Other cells model | H9C2 [46][205] | Isoproterenol | Pretreatment with Tempolcould reduce the production of reactive oxygen species and the deposition of lipid droplets and protect mitochondrial function by reducing mitochondrial swelling |
| Computational model [71][227] | Three potential dominant mechanisms are related to the effects of β-adrenergic stimulation |