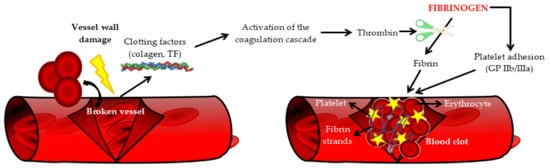
Atherosclerotic cardiovascular diseases (ASCVD), including coronary artery disease, cerebrovascular disease, and peripheral arterial disease, represent a significant cause of premature death worldwide. Biomarkers, the evaluation of which would allow the detection of ASCVD at the earliest stage of development, are intensively sought. Moreover, from a clinical point of view, a valuable biomarker should also enable the assessment of the patient’s prognosis. It has been known for many years that the concentration of fibrinogen in plasma increases, inter alia, in patients with ASCVD. On the one hand, an increased plasma fibrinogen concentration may be the cause of the development of atherosclerotic lesions (increased risk of atherothrombosis); on the other hand, it may be a biomarker of ASCVD, as it is an acute phase protein. In addition, a number of genetic polymorphisms and post-translational modifications of fibrinogen were demonstrated that may contribute to the risk of ASCVD.
- fibrinogen
- atherosclerosis
1. Fibrinogen—Physiological and Pathophysiological Aspects
| Main Pro-Atherogenic Properties of Fibrinogen | ||
|---|---|---|
|
2. Fibrinogen Molecular Modifications and Cardiovascular Risk
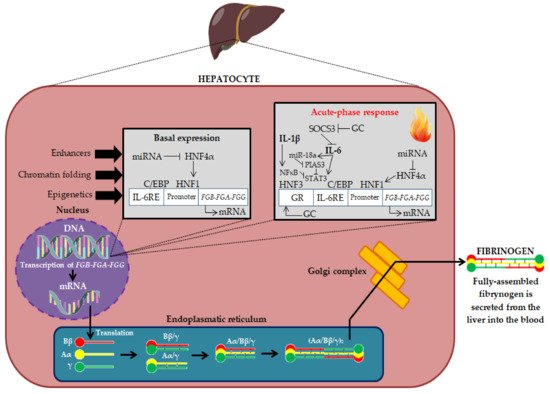
A number of molecular modifications, such as gene polymorphisms, alternative splicing and post-translational modifications, may influence the plasma concentration of fibrinogen and its biochemical properties, and consequently the risk of CVD. As mentioned earlier, fibrinogen biosynthesis begins with the expression of the three genes FGA, FGB and FGG. Many studies indicate the significant role of modifications at the stage of fibrinogen biosynthesis in shaping the risk of CVD. An alternatively-spliced form of the fibrinogen is γ′ fibrinogen (Figure 3) [67,68,69][14][15][16].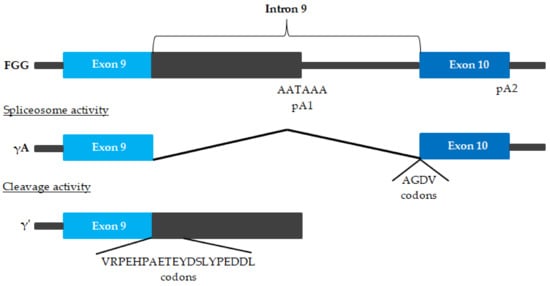
| Gene | Polymorphism/Mutation | Effect on Cardiovascular Risk | Bibliography |
|---|
| FGG | γ′ fibrinogen | ↑ myocardial infraction ↑/↔ CVD ↓ venous thromboembolism and ischemic stroke |
[67, | [ | 74, | 14 | 76,77] | ][25][30][23] |
| rs7681423 and rs1049636 | ↔ CVD | [67] | [14] | |||||
| rs2066865 | ↑ microvascular thrombosis | [75] | [31] | |||||
| FGB | -455 G/A | ↔ venous thromboembolism ↓ venous thromboembolism (Caucasians) ↑ ischemic stroke (Asian) ↔ ischemic stroke (Caucasians and children) ↑ cerebral infarction ↑ CAD ↑ cardioembolic stroke |
[84,85,86,87,88,89] | [32][33][34][35][36][37] | ||||
| -148 C/T | ↔ venous thromboembolism ↑ ischemic stroke (Asians and Caucasians) ↑ cerebral infarction ↑ CAD ↑ MACE |
[84,85,86] | [32][33][34] | |||||
| -1420 (AG + AA) and -148 (CT + TT) | ↑ lower extremity deep venous thrombosis | [95] | [26] |
References
- Pieters, M.; Wolberg, A.S. Fibrinogen and fibrin: An illustrated review. Res. Pract. Thromb. Haemost. 2019, 3, 161–172.
- Vilar, R.; Fish, R.J.; Casini, A.; Neerman-Arbez, M. Fibrin(ogen) in human disease: Both friend and foe. Haematologica 2020, 105, 284–296.
- Neerman-Arbez, M.; Casini, A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int. J. Mol. Sci. 2018, 19, 192.
- Murdaca, G.; Spanò, F.; Cagnati, P.; Puppo, F. Free radicals and endothelial dysfunction: Potential positive effects of TNF-α inhibitors. Redox Rep. 2013, 18, 95–99.
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arter. Thromb. Vasc. Biol. 2017, 37, 13–21.
- Simurda, T.; Snahnicanova, Z.; Loderer, D.; Sokol, J.; Stasko, J.; Lasabova, Z.; Kubisz, P. Fibrinogen martin: A novel mutation in FGB (Gln180Stop) causing congenital afibrinogenemia. Semin. Thromb. Hemost. 2016, 42, 455–458.
- De Vries, J.J.; Snoek, C.J.M.; Rijken, D.C.; de Maat, M.P.M. Effects of Post-Translational Modifications of Fibrinogen on Clot Formation, Clot Structure, and Fibrinolysis: A Systematic Review. Arter. Thromb. Vasc. Biol. 2020, 40, 554–569.
- Siegerink, B.; Rosendaal, F.R.; Algra, A. Genetic variation in fibrinogen; its relationship to fibrinogen levels and the risk of myocardial infarction and ischemic stroke. J. Thromb. Haemost. 2009, 7, 385–390.
- Lee, A.J.; Lowe, G.D.; Woodward, M.; Tunstall-Pedoe, H. Fibrinogen in relation to personal history of prevalent hypertension, diabetes, stroke, intermittent claudication, coronary heart disease, and family history: The Scottish Heart Health Study. Br. Heart J. 1993, 69, 338–342.
- Kannel, W.B.; D’Agostino, R.B.; Belanger, A.J. Update on fibrinogen as a cardiovascular risk factor. Ann. Epidemiol. 1992, 2, 457–466.
- Kaptoge, S.; White, I.R.; Thompson, S.G.; Wood, A.M.; Lewington, S.; Lowe, G.D.O.; Danesh, J. Associations of plasma fibrinogen levels with established cardiovascular disease risk factors, inflammatory markers, and other characteristics: Individual participant meta-analysis of 154,211 adults in 31 prospective studies: The fibrinogen studies collaboration. Am. J. Epidemiol. 2007, 166, 867–879.
- Kryczka, K.E.; Kruk, M.; Demkow, M.; Lubiszewska, B. Fibrinogen and a triad of thrombosis, inflammation, and the renin-angiotensin system in premature coronary artery disease in women: A new insight into sex-related differences in the pathogenesis of the disease. Biomolecules 2021, 11, 1036.
- Lowe, G.D.O.; Rumley, A.; Mackie, I.J. Plasma fibrinogen. Ann. Clin. Biochem. 2004, 41, 430–440.
- Lovely, R.S.; Yang, Q.; Massaro, J.M.; Wang, J.; D’Agostino Sr, R.B.; O’Donnell, C.J.; Shannon, J.; Farrell, D.H. Assessment of genetic determinants of the association of γ’ fibrinogen in relation to cardiovascular disease. Arter. Thromb. Vasc. Biol. 2011, 31, 2345–2352.
- De Willige, S.U.; Standeven, K.F.; Philippou, H.; Ariëns, R.A.S. The pleiotropic role of the fibrinogen gamma’ chain in hemostasis. Blood 2009, 114, 3994–4001.
- Farrell, D.H. Primetime for γ′. Blood 2014, 124, 1389–1390.
- Appiah, D.; Schreiner, J.P.; MacLehose, R.F.; Folsom, A.R. Association of plasma γ’ fibrinogen with incident cardiovascular disease: The atherosclerosis risk in communities (ARIC) study. Arter. Thromb. Vasc. Biol. 2015, 35, 2700–2706.
- Macrae, F.L.; Swieringa, F.; Heemskerk, J.W.M.; Ariëns, R.A.S. High fibrinogen γ’ levels in patient plasma increase clot formation at arterial and venous shear. Blood Adv. 2021, 5, 3468–3477.
- Kotzé, R.C.M.; Ariëns, R.A.S.; de Lange, Z.; Pieters, M. CVD risk factors are related to plasma fibrin clot properties independent of total and or γ’ fibrinogen concentration. Thromb. Res. 2014, 134, 963–969.
- Pieters, M.; Kotze, R.C.; Jerling, J.C.; Kruger, A.; Ariëns, R.A.S. Evidence that fibrinogen γ’ regulates plasma clot structure and lysis and relationship to cardiovascular risk factors in black Africans. Blood 2013, 121, 3254–3260.
- Rautenbach, P.H.; Nienaber-Rousseau, C.; de Lange-Loots, Z.; Pieters, M. Certain associations between iron biomarkers and total and γ’ fibrinogen and plasma clot properties are mediated by fibrinogen genotypes. Front. Nutr. 2021, 8, 720048.
- Rautenbach, P.H.; Nienaber-Rousseau, C.; Pieters, M. The association of alcohol with circulating total fibrinogen and plasma clot density is mediated by fibrinogen and FXIII genotypes. Thromb. J. 2020, 18, 35.
- Maners, J.; Gill, D.; Pankratz, N.; Laffan, M.A.; Wolberg, A.S.; de Maat, M.P.M.; Ligthart, S.; Tang, W.; Ward-Caviness, C.K.; Fornage, M.; et al. A Mendelian randomization of γ’ and total fibrinogen levels in relation to venous thromboembolism and ischemic stroke. Blood 2020, 136, 3062–3069.
- Walton, B.L.; Getz, T.M.; Bergmeier, W.; de Willige, S.U.; Wolberg, A.S. Gamma prime fibrinogen does not cause arterial thrombosis. Blood 2013, 122, 1092.
- Mannila, M.N.; Lovely, R.S.; Kazmierczak, S.C.; Eriksson, P.; Samnegård, A.; Farrell, D.H.; Hamsten, A.; Silveira, A. Elevated plasma fibrinogen gamma’ concentration is associated with myocardial infarction: Effects of variation in fibrinogen genes and environmental factors. J. Thromb. Haemost. 2007, 5, 766–773.
- Han, S.; Yang, B.; Feng, Y.; Zhao, L.; Feng, Q.; Guan, H.; Song, D.; Yin, F.; Zhuang, L. The correlation between FGB promoter polymorphism and clotting function in patients with idiopathic lower extremity deep venous thrombosis. Clin. Appl. Thromb. Hemost. 2021, 27, 1076029620967108.
- Simurda, T.; Brunclikova, M.; Asselta, R.; Caccia, S.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Genetic variants in the FGB and FGG genes mapping in the beta and gamma nodules of the fibrinogen molecule in congenital quantitative fibrinogen disorders associated with a thrombotic phenotype. Int. J. Mol. Sci. 2020, 21, 4616.
- Simurda, T.; Caccia, S.; Asselta, R.; Zolkova, J.; Stasko, J.; Skornova, I.; Snahnicanova, Z.; Loderer, D.; Lasabova, Z.; Kubisz, P. Congenital hypofibrinogenemia associated with a novel heterozygous nonsense mutation in the globular C-terminal domain of the γ-chain (p.Glu275Stop). J. Thromb. Thromb. 2020, 50, 233–236.
- Simurda, T.; Casini, A.; Stasko, J.; Hudecek, J.; Skornova, I.; Vilar, R.; Neerman-Arbez, M.; Kubisz, P. Perioperative management of a severe congenital hypofibrinogenemia with thrombotic phenotype. Thromb. Res. 2020, 188, 1–4.
- Appiah, D.; Heckbert, S.R.; Cushman, M.; Psaty, B.M.; Folsom, A.R. Lack of association of plasma gamma prime (γ’) fibrinogen with incident cardiovascular disease. Thromb. Res. 2016, 143, 50–52.
- Drizlionoka, K.; Zariņš, J.; Ozolina, A.; Ņikitina-Zaķe, L.; Mamaja, B. Polymorphism rs2066865 in the fibrinogen gamma chain (FGG) gene increases plasma fibrinogen concentration and is associated with an increased microvascular thrombosis rate. Medicina 2019, 55, 563.
- Li, D.; Zhang, X.; Huang, H.; Zhang, H. Association of β-fibrinogen polymorphisms and venous thromboembolism risk: A PRISMA-compliant meta-analysis. Medicine 2019, 98, e18204.
- Luo, H.; Li, X.; Jiang, A.; Zhang, B.; Bi, P.; Dong, Y.; Guo, Y. Associations of β-fibrinogen polymorphisms with the risk of ischemic stroke: A meta-analysis. J. Stroke Cerebrovasc. Dis. 2019, 28, 243–250.
- Canseco-Avila, L.M.; Lopez-Roblero, A.; Serrano-Guzman, E.; Aguilar-Fuentes, J.; Jerjes-Sanchez, C.; Rojas-Martinez, A.; Ortiz-Lopez, R. Polymorphisms -455G/A and -148C/T and fibrinogen plasmatic level as risk markers of coronary disease and major adverse cardiovascular events. Dis. Markers 2019, 2019, 5769514.
- Gu, L.; Wu, G.; Su, L.; Yan, Y.; Long, J.; Tan, J.; Liang, B.; Guo, X.; Huang, G. Genetic polymorphism of β-fibrinogen gene-455G/A can contribute to the risk of ischemic stroke. Neurol. Sci. 2014, 35, 151–161.
- Gu, L.; Liu, W.; Yan, Y.; Su, L.; Wu, G.; Liang, B.; Tan, J.; Huang, G. Influence of the β-fibrinogen-455G/A polymorphism on development of ischemic stroke and coronary heart disease. Thromb. Res. 2014, 133, 993–1005.
- Hu, X.; Wang, J.; Li, Y.; Wu, J.; Qiao, S.; Xu, S.; Huang, J.; Chen, L. The β-fibrinogen gene 455G/A polymorphism associated with cardioembolic stroke in atrial fibrillation with low CHA2DS2-VaSc score. Sci. Rep. 2017, 7, 17517.
- Cronjé, H.T.; Nienaber-Rousseau, C.; Zandberg, L.; Chikowore, T.; de Lange, Z.; van Zyl, T.; Pieters, M. Candidate gene analysis of the fibrinogen phenotype reveals the importance of polygenic co-regulation. Matrix Biol. 2017, 60–61, 16–26.
- Cronjé, H.T.; Nienaber-Rousseau, C.; Zandberg, L.; de Lange, Z.; Green, F.R.; Pieters, M. Fibrinogen and clot-related phenotypes determined by fibrinogen polymorphisms: Independent and IL-6-interactive associations. PLoS ONE 2017, 12, e0187712.
- Titov, B.V.; Barsova, R.M.; Martynov, M.Y.; Nikonova, A.A.; Favorov, A.V.; Gusev, E.I.; Favorova, O.O. Polymorphic variants of genes encoding interleukin-6 and fibrinogen, the risk of ischemic stroke and fibrinogen levels. Mol. Biol. 2012, 46, 93–102, (Article in Russian, Only Abstract in English).
- Ward-Caviness, C.K.; de Vries, P.S.; Wiggins, K.L.; Huffman, J.E.; Yanek, L.R.; Bielak, L.F.; Franco, G.; Xiuqing, G.; Marcus, E.K.; Tim, K.; et al. Mendelian randomization evaluation of causal effects of fibrinogen on incident coronary heart disease. PLoS ONE 2019, 14, e0216222.