MicroRNAs (miRNA) are small non-coding RNAs that are 20–23 nucleotides in length, functioning as regulators of oncogenes or tumor suppressor genes. They are molecular modulators that regulate gene expression by suppressing gene translation through gene silencing/degradation, or by promoting translation of messenger RNA (mRNA) into proteins. Circulating miRNAs have attracted attention as possible prognostic markers of cancer, which could aid in the early detection of the disease. Epithelial to mesenchymal transition (EMT) has been implicated in tumorigenic processes, primarily by promoting tumor invasiveness and metastatic activity; this is a process that could be manipulated to halt or prevent brain metastasis. Studies show that miRNAs influence the function of EMT in glioblastomas. Thus, miRNA-related EMT can be exploited as a potential therapeutic target in glioblastomas.
- microRNA
- EMT
1. Introduction
2. Epithelial Mesenchymal Transition in Glioblastoma
During EMT, epithelial cells develop a mesenchymal phenotype and properties including migration. Cancer cells acquire these abilities and migrate to distant sites away from the primary tumor to establish metastatic foci. The process of EMT and the resultant migratory abilities are also associated with drug resistance. A number of processes contributing to glioma EMT have been described. The induction of these processes in glioblastomas differ from those observed in epithelial cancers because of the absence of the basement membrane [7][8][9,10]. The basement membrane is a specialized extracellular matrix that forms the base for endothelial and epithelial cells. It plays a pivotal role in maintaining the structural integrity, exchanging biochemical signals, and regulating cellular function [9][10][11,12]. Typically, for tumor cells to migrate to other parts of the body, they need to detach from the basement membrane. In the case of gliomas, glial cells are the most abundant cells in the brain, and provide protective and structural support [11][13]. A type of glial cell, referred to as astrocyte, becomes activated and surrounds the tumor stroma, thus keeping cancer cells and the cluster intact. These cells can undergo EMT processes induced by cancer cells and EMT-associated transcription factors [12][14] (Figure 1).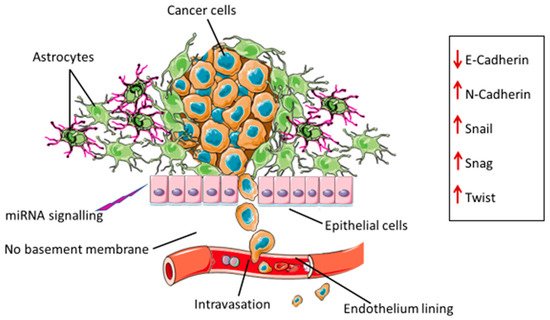
3. Drivers of EMT in Glioblastoma
3.1. Extracellular Vesicles (EVs)
3.2. Transforming Growth Factor β Signaling Pathway
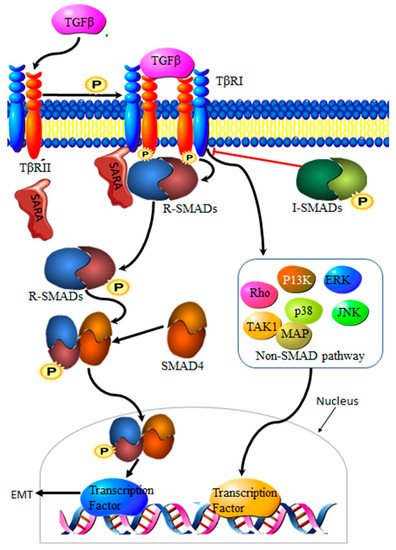
Transforming growth factor-β (TGF-β) is a family of multi-functional growth factors involved in cell proliferation, differentiation and apoptosis. During cancer development, TGF-β suppresses cancer growth via cell cycle arrest and apoptosis, but supports cancer progression at a later stage of the disease by promoting invasion, increasing migration capabilities and drug resistance [18][23]. The TGF-β complex consists of surface receptor type I (TβRI) and type II (TβRII) transmembrane serine/threonine kinases. Signaling is initiated by the binding of TGF-β to TβRII and TβRI receptors on the cell surface, resulting in a heterocomplex. Phosphorylation by TβRII activates TβRI, resulting in the recruitment and phosphorylation of Smad proteins 2 and 3, collectively known as R-Smads. Translocation to the nucleus requires the binding of R-Smads to Smad 4. The resultant R-Smads/Smad 4 complex will then induce the transcription of target proteins [19][24] (Figure 2). Aberrant TGF-β signaling pathways contribute to cancer development [20][25], making TGF-β an important driver of EMT in cancers.
3.3. Autophagy
3.4. MiRNAs
The expression signatures of a number of miRNAs have been identified as either instigators or inhibitors of EMT in glioblastomas. Zhang et al. assessed the clinical importance of EMT in glioma using independent glioma datasets, GSE16011, Rembrandt and The Cancer Genome Atlas (TCGA) program. The study found 19 miRNA expression signatures that are positively correlated with EMT-driven gliomas, with the most prominent being miR-223 [5][7]. Blocking the miR-223/PAX6 pathway improved sensitivity to chemotherapeutic treatment with temozolomide in glioblastomas, indicating the role of miR-223 in drug resistance [25][43]. The PI3K/Akt signaling pathway is associated with EMT-related disease severity in glioblastomas. This pathway has been shown to act in collaboration with other EMT-related signaling pathways, including mTOR [26][44], CXCR4 signaling [27][45] and LASP1 [28][46].4. miRNA-EMT-Related Cancer Cell Invasion and Metastasis
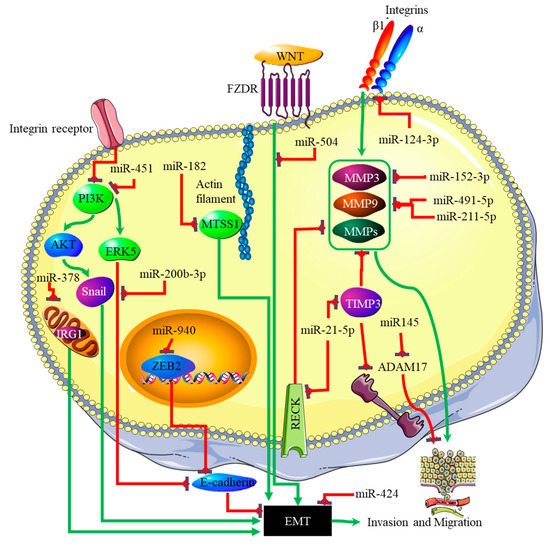
The link between miRNA expression signatures, EMT and, ultimately, cancer metastasis has long been established [29][60]. Several miRNA signatures are involved in EMT-associated mechanisms of invasion and metastasis in glioblastomas. Studies on miR-125a-5p found its expression to suppress the mesenchymal capabilities of glioblastoma cells, thus inhibiting EMT [30][31][61,62]. Recently, Nan et al. showed that miR-451 expression is able to suppress EMT and metastasis by blocking the PI3K/Akt/Snail signaling pathway through the activation of calcium-binding protein 39 (CAB39) in gliomas [32][63]. The expression of miR-451 was associated with the inhibition or reversal of the EMT processes in cancers [33][64]. Contrary to this finding, miR-451 expression plays different roles in gliomas, as it was associated with the proliferation of cancer cells by suppressing the CAB39/AMPK/mTOR pathway, and increased metastatic potential, which takes place via the activation of the Rac1/cofilin pathway [34][65]. The increased expression of miR-200b-3p prompted E-cadherin (essential for maintaining the structural integrity of epithelial cells) by the deactivation of extracellular signal-regulated kinase 5 (ERK5), resulting in reduced glioma cell proliferation and mesenchymal capabilities [35][41]. mR-424 showed promising results as a potential prognostic molecular marker and therapeutic target by blocking EMT processes and the resultant metastatic capabilities by targeting the kinesin-like protein KIF23 in human glioma [36][66]. The same effect was observed with miR-378, which inhibited EMT by targeting cis-aconitate decarboxylase (IRG1) in gliomas [37][67], miR-139-5p by targeting the notch1 oncogene in gliomas [38][68], miR-181a by targeting ZBTB33 expression in glioma cells [39][69], miR-623 by targeting TRIM-44 [40][70], miR-940 by targeting ZEB2 [41][71], and miR-7, which targeted T-Box 2 in glioblastoma [42][72]. In addition to these miRNAs, miR-182-targeted MTSS1 enhanced TGF-β1-related EMT in gliomas [43][73], and miR-504 inhibited EMT in glioblastomas by targeting the Wnt receptor FZD7/β-catenin pathway. The phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase (PTEN) tumor suppressor inhibits PI3K by dephosphorylating it. The expression of PTEN is inhibited through the action of three separate miRNAs. These are miR-17-5p [44][74], miR-23a-3p [45][75] and miR-26a-5p [46][76]. The p53 tumor suppressor is, itself, downregulated by miR-10b-5p. This miRNA also downregulates the tumor suppressor and cell cycle regulator p16 [47][77]. At the same time, the negative regulator of p53, MDM2, is upregulated in glioblastoma by miRNAs such as miR-32-5p, miR-25-3p and miR-17-3p [48][78]. The RB1 tumor suppressor is downregulated by the miRNA miR-28-5p. At the same time, the expression and activity of oncogenes that promote cell cycle progression are downregulated by various miRNAs in glioblastoma; these include miR-124-3p suppressing cyclin-dependent kinase (CKD) 4 [49][79], and miR-491-5p, miR-491-3p and miR-138-5p [50][80] suppressing CKD6. Finally, the proliferative activity of cyclin D is downregulated by miR-195-5p [51][81]. A decreased expression ratio of the miRNA miR-504/FZD7 was shown to be a potential molecular marker for identifying the mesenchymal subtype in glioblastoma [52][82]. The actions of the miRNAs discussed above are summarized in Figure 34.
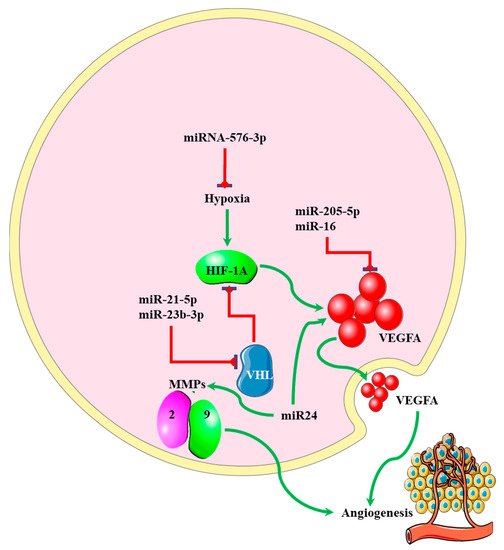
5. miRNA-EMT-Related Angiogenesis
MicroRNAs regulating angiogenesis in cancers have been dubbed angiomiRNAs [53][54][85,86]. Several angiomiRNAs have been identified in glioblastomas [55][87], with some originating from glioblastoma-derived EVs [56][88]. However, studies indicating the relationship between angiomiRNAs and the EMT process in glioblastomas are limited. Dai et al. found that upregulated miR-24 expression induced the expression of the angiogenic markers VEGF and, TGF-β, and matrix metalloproteinases (MMP)-2 and -9, resulting in increased glioblastoma cell proliferation and development [57][89] (Figure 45). Increased expression of miR-16 in glioblastoma inhibited EMT processes by targeting polycomb complex protein BMI-1, and reducing the expression levels of the angiogenic markers VEGF-A and VEGF-C [58][90]. miR-576-3p was shown to inhibit EMT and the angiogenic properties of hypoxia-treated glioma cells by targeting HIF-1α [59][91] (Figure 45).