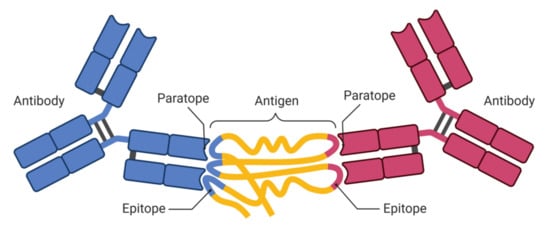
The unreliability issue in predicting BCEs due to amino acid scales has been mitigated using ML algorithms. To differentiate BCEs from non-epitopes, ML algorithms have been trained on feature vectors extracted from BCEs. A few methods
, as illustrated in Table 3, based on ML include ABCpred
[48], BCPREDS
[49], LBtope
[50], SVMtrip
[51], and BepiPred
[52]. It has been reported that methods based on ML techniques outperform the techniques based on amino acid scales
[53]. Conformational BCEs constitute the majority portion; however, their prediction is lagging behind that of linear types due to two main reasons. Firstly, their prediction necessitates knowledge of the 3D protein structure. Only a limited percentage of proteins have 3D information
[54]. Secondly, extracting conformational epitopes for specific antibody synthesis from a protein context is a difficult process that requires the use of appropriate scaffolds for epitope grafting. Therefore, their prediction thus far is of less relevance for EBPV design.
4. Predicting SARS-CoV-2 Epitopes
Coronaviruses belong to the family
Coronaviridae, the enveloped viruses having a large single-stranded RNA genome whose length ranges from 26 to 32 kilobases
[55]. In
[56], by Lineburg and colleagues, it has been found that, among 26 viral proteins of SARS-CoV-2, a few proteins on its surface, such as the spike protein (S), are more variable, while others are more conserved and internal, such as the nucleocapsid protein (N). It has been found that the spike protein (S) is responsible for activating cytotoxic CD8+ T cells and hence is considered an ideal vaccine target.
The infection caused by SARS-CoV-2 elicits both adaptive and innate arms of immunity
[57]. In general, antigen-presenting cells recognize viruses. Once T-cell activation happens, CD4+ T cells mainly differentiate into effector cells, which produce cytokines and chemokines; cytotoxic CD8+ T cells, on the other hand, are key players in the immune response to viral infection, as they participate directly in viral clearance
[58]. It has been demonstrated that T cells, apart from targeting the structural proteins of coronaviruses, are also responsible for lung immunopathological damage due to SARS-CoV and MERS-CoV
[59][60]. Thus, in the case of SARS-CoV-2, the major focus has been on identifying viral T-cell epitopes presented on human leukocyte antigens (HLA)
[61][62] (
Table 24).
Table 24.
Existing ML methods used in SARS-CoV-2 epitope prediction.
| Sr. No. |
Method Name |
Usage |
| 01 |
NetMHC | [63] |
To predict HLA I class or CD8+ SARS-CoV-2 T-cell epitopes |
| 02 |
NetMHCpan | [64] |
| 03 |
NetCTLpan_1.1 | [65] |
| 04 |
NetMHC_4.0 | [66] |
| 05 |
HLAthena | [67] |
| 06 |
MHCflurry | [68] |
| 07 |
NetHMCII_2.3 | [69] |
To predict HLA II class or CD4+ SARS-CoV-2 T-cell epitopes |
| 08 |
NetMHCIIpan_3.0 | [70] |
| 09 |
NetMHCIIpan_4.0 | [71] |
| 10 |
NeonMHC2 | [72] |
| 11 |
MARIA | [73] |
A few techniques listed in
Table 24 have “pan” as a suffix, which indicates an ability to predict the binding of HLA peptides for a huge collection of the alleles inside a particular HLA type, including those not present in the training dataset
[72]. A few studies have also used algorithms specific to HLA-I, namely Net_Chop
[74] and NetCTL1.2
[75], where extra- and intracellular variables responsible for the presentation of HLA antigens were integrated to improve the prediction accuracy of the binding of peptide HLA. The methods NetCTL-1.2
[75] and NetChop
[74] have also been utilized in a few studies, where extra- and intracellular variables have been integrated, which are responsible for presenting HLA antigens. It is essential to mention here that almost all modern T-cell epitope prediction systems use ANNs. A few early ones (such as RANKPEP
[76] and CTLPred
[41]) used a different ML approach, support vector machines (SVM). The spike proteins in the original virus bind to the ACE2 receptor on human cells. It has been reported in
[77] that the D614G mutation alters the genetic code of the spike protein of SARS-CoV-2, where a change in a single amino acid takes place, and most of the COVID-19 vaccines are based on this spike protein. Due to this mutation, the virus spreads faster and the spikes become more stable than those in the original virus. As a result, more functional spikes are available to bind to ACE2 receptors, making the virus more infectious. Crooke et al.
[78] developed a computational model using various open-source algorithms and web-based tools to analyze the SARS-CoV-2 proteome so as to identify antigenic and putative T-cell and B-cell epitopes as potential vaccine targets. After using a set of stringent selection criteria to filter out the peptide epitopes, the study discovered 41 T-cell epitopes (5 HLA class I, 36 HLA class II) and six B-cell epitopes that have the potential to serve as primary targets for epitope-based peptide vaccine development against SARS-CoV-2.
5. Future Research Directions in T- and B-Cell Epitope Prediction
By now, it is clear that the key to designing an EBPV is the identification of BCEs and TCEs
[79][80]. Several studies have been performed to predict BCEs and TCEs, as illustrated in
Table 1.
The methFo
ds used to predict SARS-CoV-2 epitopes are listed in Table 2; again, these predir eac
t only th
e peptide-binding capacity. This is a limitation with these methods; instead of predicting the binding capability of a peptide, predicting epitopes deterministically is desired. Because viruses continue to mutate, as with SARS-CoV-2, existing vaccines may prove to be somewhat less effective against new variants. Either the vaccine’s composition has to be changed or a new vaccine needs to be developed to protect against these variants [81]. Tim study, we have mentioned our opinions in terms of their stre
being
the critical factor, EBPVs can be a great solution. Based on the research conducted, EBPVs are highly recommended vaccines and should be considered in the quest for the rapid development of protective vaccines. Below, we mention the future research directions for epitope prediction as predicting epitopes is a sensitive task and needs due attention in order to improve it.
1. The mths and limitations. Apart from these studies, severa
jority ofl current state-of-the-art approaches estimate a peptide’s binding capability. These approaches struggle to predict deterministically whether a given peptide is an epitope or not. CTLpredtools and methods are [41], one of the serav
ers, opera
tes in this category; however, it is limited to peptides that are up to 9 mers in length. To circumvent the limitations of the previous approaches, a direct method of predicting epitopes is sought. Furthermore, the technique should be capable of pilable online for free to predict
ing variable-length peptides with a length greater than 9 mers.
2. Current state B- and T-
of-thce
-art MLll epitope
prediction approaches rely heavily on just a few classifiers, including ANNs, SVMs, s, as illustrated in Table 2 and
Hidden Markov models (HMM) [82] Table 3. Th
ere
are other robust classifiers available that can be utilized to achieve even more promising results, including decision trees (DT), random forest (RF), convolutional neural networks (CNNs), and AdaBoost [83]. In the literature surveyed, ANNmethods used to predict SARS-CoV-
based2 models constitute the majority of the eepitope
prediction methods. However, relying on ANNs only is not safe. ANNs suffer from a hardware dependencys are listed in Table 4; a
s they require processors with parga
llel processing power in accordance with their structure [84]. Because epitopin, these predict
ion is such a delicate task, the ANN’s behavior is occasionally unexplainable. When an ANN generates a probing solution, it does not explain why or how it was generated, which reduces the trust in the network [84]. However, to have highonly the peptide-
performing models and rob
ust models for applications such as the healthcare domain, explainable ML can be explored, which is in its initial stage and remains an open issue [85]. Gagniuinding c
et a
l. have proposed a spectral-based forecast model as an alternative to the classical ANN. In their experiment, the ANN categorized the collection of data fairly but failed to reveal any useful information about the evolution of a subject over time. In this regard, forecasts based on Markov chains or traditional statistical methodologies have produced more trustworthy outcomes in the biology and medicine domains. The proposed novel method of analysis based on spectral forecasts outperformed the classical ANNs [86].
3. Moreover, ipacity. This is a limitation with these methods; instead of
relying on predicti
ons by a single model, we can combine several robust classifiers, called an ensemble model. Ensemble learning (EL) is a powerful technique for boosting the model accuracy by combining a number of base classifiers [87]. Such a technique has considerably better generalization cang the binding capability
than itsof individual counterparts. Indeed, EL is appealing because it can elevate weak learners (also known as base classifiers), which are marginally better than random guesses, to strong learners, which can make accurate forecasts [88]. The base classa peptide, predicting epitopes determi
fiers vote for a n
ew data instance, and, based on the majority of votes, a class label is returned. An ensemble model can be created by training homogeneous base models on different subsets of the training set or heterogeneous base models using the same training dataset. The main three types of ensembling techniques are bagging, boosting, and stacking. Multiple base learners (homogenous) can be integrated in bagging using different sub-samples from the same dataset [89]. The final predictiistically is desired. Because viruses continue to mutate, as with SARS-CoV-2, existing vaccines may prove to
n is obtained by taking the average prediction from multiple base learners. In boosting, base learners are added sequentially, and the predictions reported by previous learners are corrected. The final output is decided by taking the weighted average of all the predictions [89]. On the other hbe somewhat less effective against new varian
d, st
acking involves fitting heterogeneous base learners on the same dataset [89] and then using another learner to learn how to best s. Either the vacc
ombine
all the predictions. Moreover, while dealing with complex data, such as high-dimensional, imbalanced, noisy data, etc., traditional ML algorithms may fail to produce satisfactory results. The reason for this is that, for these methods, it is difficult to capture various attributes and the underlying layout of the data. Ensemble learning aims to combine data modeling, data fusion, and data mining into a cohesive framework [90] To conc’s composition has to be changed or a new vaccine needs to be devel
ude, the main reaso
ns for employing ensemble learning in epitope prediction are as follows:
-
Performance: An ensemble can outperform any single contributing model in terms of prediction and performance [91].
-
Robustness: An ensemble narrows the spread or dispersion of predictions and improves model robustness and reliability [91].
4. In ped to prot
he
literature surveyed, not all physicochemical properties of amino acids have been utilized to extract features from peptide sequences. To have a robust epitope prediction system in place, additional physicochemical propertiect against these variants
need to be explored [92][9381].
5. T
he exi
sting ML-based methods for epitope prediction have been assessed using metrics such as accuracy and area under the curve (AUC). However, other confusion matrix-based performance metrics such as Gini, specificity, sensitivity, F-score, kappa, Matthews correlation coefficient (MCC), and precision, etc., can be utilized to analyze the performance of the model in a better way.
6. Conclusions
Prediction of T- and B-cell epitopes can play a game-changing role in the me being the critical factor, EBPV
des
ign process, as well as in disease diagnosis. In this study, a review of various existing studies for epitope prediction has been provided. Moreover, a review has been provided for the state of-the-art ML-based tools that are available online and free to use for researchers working in vaccine design. The COVID-19 pandemic, caused by the SARS-VoV-2 virus, has resulted in a dramatic loss of human life worldwide and poses an unprecedented challenge to public health, food systems, and the workplac can be a great solution. Based on the research conducted, EBPVs are
[94]. Accordingly, a special emph
asi
s has been placed on highlighting and analyzing various ML-based methods that have been proposed and used for predicting epitopes of SARS-CoV-2 for EPBV design in order to contain the COVID-19 pandemic. However, it is important to mention here that the application of epitope prediction tools/methods to SARS-CoV-2 presented in this review is not satisfactorily developed, and only a few them have been applied for SARS-CoV-2 epitope prediction. Another reason to place special emphasis on SARS-CoV-2 is that the EPBV design approach seems to be a promising alternative in order to quickly design new vaccines against different variants of the virus as it continues to mutate [95]. Based ghly recommended vaccines and should be considered in the quest fo
n the var
ious state-of-the-art ML methods discussed, future research directions for epitope prediction have been presented. From the literature reviewed, it has been observed that focus has been given to peptide-binding capability prediction instead of deterministically predicting whether a peptide is an epitope or not. In addition, the majority of the ML-based prediction models are based on a single classifier. However, instead of relying on a single model, several robust classifiers can be combined into an ensemble model in order to enhance the epitope prediction accuracy. To conclude, it is important to m the rapid development of protective vaccines. Below, we mention th
at the prediction of T-cell epitopes is much more reliable and advanced as compared to the prediction of B-cell epitopes. Moreover, ife future research directions for epitope
s are predict
ed efficiently using computational approaches (ML-based methods), they can be used as futuristic vaccine candidates with fewer side effects compared to conventional vaccine designs subjected to in vitro and in vivo scientific assessments. The technology developed would help the broad scientific community working in vaccine development to save time in screening the active epitope candidates against the inactive ones. In conclusion, it is relevant to provide a review of the existing ML-based state-of-the-art methods for TCE and BCE prediction because EBPVs have significant potential and should be considered in the quest for the rapid development of a protective vaccine against a pathogen, specifically for SARS-CoV-2, as there is a strong likelihood that the virus will mutate further. This will also stimulate continuing research efforts for the EBPV design procession as predicting epitopes is a sensitive task and needs due attention in order to improve it.
1.
The majority of current state-of-the-art approaches estimate a peptide’s binding capability. These approaches struggle to predict deterministically whether a given peptide is an epitope or not. CTLpred [41], one of the servers, operates in this category; however, it is limited to peptides that are up to 9 mers in length. To circumvent the limitations of the previous approaches, a direct method of predicting epitopes is sought. Furthermore, the technique should be capable of predicting variable-length peptides with a length greater than 9 mers.
2.
Current state-of-the-art ML epitope prediction approaches rely heavily on just a few classifiers, including ANNs, SVMs, and Hidden Markov models (HMM) [82]. There are other robust classifiers available that can be utilized to achieve even more promising results, including decision trees (DT), random forest (RF), convolutional neural networks (CNNs), and AdaBoost [83]. In the literature surveyed, ANN-based models constitute the majority of the epitope prediction methods. However, relying on ANNs only is not safe. ANNs suffer from a hardware dependency as they require processors with parallel processing power in accordance with their structure [84]. Because epitope prediction is such a delicate task, the ANN’s behavior is occasionally unexplainable. When an ANN generates a probing solution, it does not explain why or how it was generated, which reduces the trust in the network [84]. However, to have high-performing models and robust models for applications such as the healthcare domain, explainable ML can be explored, which is in its initial stage and remains an open issue [85]. Gagniuc et al. have proposed a spectral-based forecast model as an alternative to the classical ANN. In their experiment, the ANN categorized the collection of data fairly but failed to reveal any useful information about the evolution of a subject over time. In this regard, forecasts based on Markov chains or traditional statistical methodologies have produced more trustworthy outcomes in the biology and medicine domains. The proposed novel method of analysis based on spectral forecasts outperformed the classical ANNs [86].
3.
Moreover, instead of relying on predictions by a single model, we can combine several robust classifiers, called an ensemble model. Ensemble learning (EL) is a powerful technique for boosting the model accuracy by combining a number of base classifiers [87]. Such a technique has considerably better generalization capability than its individual counterparts. Indeed, EL is appealing because it can elevate weak learners (also known as base classifiers), which are marginally better than random guesses, to strong learners, which can make accurate forecasts [88]. The base classifiers vote for a new data instance, and, based on the majority of votes, a class label is returned. An ensemble model can be created by training homogeneous base models on different subsets of the training set or heterogeneous base models using the same training dataset. The main three types of ensembling techniques are bagging, boosting, and stacking. Multiple base learners (homogenous) can be integrated in bagging using different sub-samples from the same dataset [89]. The final prediction is obtained by taking the average prediction from multiple base learners. In boosting, base learners are added sequentially, and the predictions reported by previous learners are corrected. The final output is decided by taking the weighted average of all the predictions [89]. On the other hand, stacking involves fitting heterogeneous base learners on the same dataset [89] and then using another learner to learn how to best combine all the predictions. Moreover, while dealing with complex data, such as high-dimensional, imbalanced, noisy data, etc., traditional ML algorithms may fail to produce satisfactory results. The reason for this is that, for these methods, it is difficult to capture various attributes and the underlying layout of the data. Ensemble learning aims to combine data modeling, data fusion, and data mining into a cohesive framework [90] To conclude, the main reasons for employing ensemble learning in epitope prediction are as follows:
-
Performance: An ensemble can outperform any single contributing model in terms of prediction and performance [91].
-
Robustness: An ensemble narrows the spread or dispersion of predictions and improves model robustness and reliability [91].
4.
In the literature surveyed, not all physicochemical properties of amino acids have been utilized to extract features from peptide sequences. To have a robust epitope prediction system in place, additional physicochemical properties need to be explored [92][93].
5.
The existing ML-based methods for epitope prediction have been assessed using metrics such as accuracy and area under the curve (AUC). However, other confusion matrix-based performance metrics such as Gini, specificity, sensitivity, F-score, kappa, Matthews correlation coefficient (MCC), and precision, etc., can be utilized to analyze the performance of the model in a better way.
6. Conclusions
Prediction of T- and B-cell epitopes can play a game-changing role in the EBPV design process, as well as in disease diagnosis. In this study, a review of various existing studies for epitope prediction has been provided. Moreover, a review has been provided for the state of-the-art ML-based tools that are available online and free to use for researchers working in vaccine design. The COVID-19 pandemic, caused by the SARS-VoV-2 virus, has resulted in a dramatic loss of human life worldwide and poses an unprecedented challenge to public health, food systems, and the workplace [94]. Accordingly, a special emphasis has been placed on highlighting and analyzing various ML-based methods that have been proposed and used for predicting epitopes of SARS-CoV-2 for EPBV design in order to contain the COVID-19 pandemic. However, it is important to mention here that the application of epitope prediction tools/methods to SARS-CoV-2 presented in this review is not satisfactorily developed, and only a few them have been applied for SARS-CoV-2 epitope prediction. Another reason to place special emphasis on SARS-CoV-2 is that the EPBV design approach seems to be a promising alternative in order to quickly design new vaccines against different variants of the virus as it continues to mutate [95]. Based on the various state-of-the-art ML methods discussed, future research directions for epitope prediction have been presented. From the literature reviewed, it has been observed that focus has been given to peptide-binding capability prediction instead of deterministically predicting whether a peptide is an epitope or not. In addition, the majority of the ML-based prediction models are based on a single classifier. However, instead of relying on a single model, several robust classifiers can be combined into an ensemble model in order to enhance the epitope prediction accuracy. To conclude, it is important to mention that the prediction of T-cell epitopes is much more reliable and advanced as compared to the prediction of B-cell epitopes. Moreover, if epitopes are predicted efficiently using computational approaches (ML-based methods), they can be used as futuristic vaccine candidates with fewer side effects compared to conventional vaccine designs subjected to in vitro and in vivo scientific assessments. The technology developed would help the broad scientific community working in vaccine development to save time in screening the active epitope candidates against the inactive ones. In conclusion, it is relevant to provide a review of the existing ML-based state-of-the-art methods for TCE and BCE prediction because EBPVs have significant potential and should be considered in the quest for the rapid development of a protective vaccine against a pathogen, specifically for SARS-CoV-2, as there is a strong likelihood that the virus will mutate further. This will also stimulate continuing research efforts for the EBPV design process.