The recently discovered clustered regularly interspaced short palindromic repeats-associated protein (CRISPR-Cas) system is involved in the natural protection of prokaryotes from foreign DNA invasion
[13][14][15][84,85,86]. CRISPR-Cas can be used to edit phage genomes, thereby conferring the advantage of specific production of the expected recombinant phages, with the removal of Cas protein inhibiting the production of wild-type phages. In recent years, the CRISPR-Cas system has been used for genome editing in several organisms, including phages. In general, the CRISPR-Cas system contains two important components: Cas protein and CRISPR RNA (crRNA). CRISPR-Cas for editing the phage genome involves three steps: (a) adaptation, (b) crRNA biogenesis, and (c) interference
[16][87]. In the adaptation step, a short foreign nucleotide sequence (30–40 bp) called the “spacer or tracer” sequence is integrated with CRISPR loci of partially palindromic DNA repeats. In the crRNA biogenesis step, the spacer and partially palindromic DNA repeats are transcribed into crRNA. In the interference step, the crRNA combines with one or more CRISPR associated proteins (Cas) to form a complex called a “protospacer”, which will recognize the DNA and finally degrade it. Based on Cas protein function, CRISPR-Cas systems are classified into six types I to VI, with each of them having several subtypes based on genetic diversity. According to the current scenario, these six types belong to two major classes: the class 1 system encodes multiple Cas proteins involved in types I, III, and IV, and the class 2 system encodes a single Cas protein function to degrade target DNA in type II, V, and VI. In recent years, types I, II, and III CRISPR systems have been mainly used for phage genome editing
[17][88]. The class 1 subtype I-E CRISPR-Cas system from
Vibrio cholerae and
E. coli and the class 2 subtype II-A system from
Streptococcus thermophilus have been used to edit the genome of lytic phages. The CRISPR-Cas system was first used to delete gene 1.7 of T7 phage
[18][89]. In the same year, the class 2 subtype II-A system from
S. thermophilus was used for the first time
[19][90]. In 2016, the
V. cholerae phage was edited with a class 1 subtype I-E CRISPR-Cas system, which contains both donor DNA and the CRISPR-Cas gene, to successfully obtain recombinant phages
[20][91]. The CRISPR-Cas9 protein system belongs to the type II CRISPR system, which requires a protospacer-adjacent motif (PAM) site to degrade foreign DNA
[21][22][23][92,93,94]. The
Streptococcus pyogenes Cas9 (SpCas9) protein has a special programmed PAM site containing three base pairs (5′-NRG-3′) to cleave target foreign DNA
[24][25][95,96]. The sequence 5′-NRG-3, where N = any nucleotide A, T, G, or C, and R = G or A, presents a range of possible SpCas9 PAM sites: AGG, TGG, GGG, CGG, AAG, TAG, GAG, and CAG.
SpCas9 targets and cleaves DNA with the help of the PAM site and spacer sequence already encoded in crRNA. The CRISPR-Cas system with SpCas9 has been used to edit several organisms, including phage genomes. When using this system, Cas9, crRNA, and tracr-RNA (trans-activating crRNA) were simultaneously cloned into the same plasmid
[26][27][28][97,98,99]. When the crRNA and tracr-RNA are fused to generate a single guide RNA (sgRNA), the properly prepared highly active sgRNA yields positive results. After the
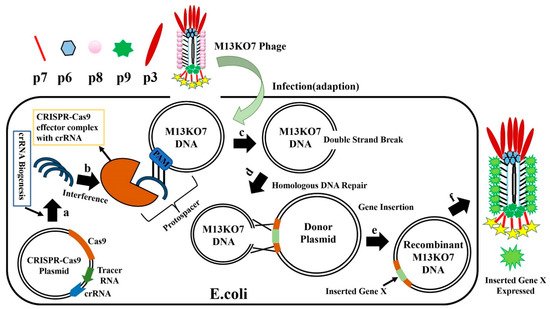
E. coli with CRISPR-Cas9-sgRNA plasmid is infected with M13KO7 phage (
Figure 10), the CRISPR-Cas9 plasmids synthesize crRNA and Cas9 protein, forming the effector Cas9-sgRNA complex. This complex has an ability to bind to the M13KO7 phage DNA at the PAM site and create a double-strand break. The broken M13KO7 phage DNA could then be repaired by the donor plasmid with Gene X in the middle of the short (40–60 bp) homologous sequence of gVIII (excluding PAM site) based on the DNA repair pathways of nonhomologous end joining or homologous DNA repair. After this repair pathway, gene insertion results in the production of the recombinant M13KO7 phage with the gene X peptide at the pVIII N-terminal. Unfortunately, a weak sgRNA may lead to false-positive results. The variable activity of crRNA has already been observed in Type I-E and Type IA CRISPR-Cas systems. Since the exact distribution of sgRNA activity in phages is still highly unknown, the prediction of high activity sgRNA in phages currently uses the sgRNA prediction tool based on a eukaryotic dataset
[29][30][31][100,101,102]. Conventional recombination technology is highly expensive and time consuming for gene editing, which requires a few rounds of PCR, restriction enzyme digestion, and ligation
[32][103]. These steps have been eliminated by the introduction of oligonucleotide recombination of the template through CRISPR-Cas systems, but the synthesis of long oligonucleotides still has limitations
[33][104]. In 2018, the CRISPR-Cas system from
Listeria monocytogenes was used for
Listeria phage genome editing
[34][105]. Later, the Type III CRISPR-Cas10 system was also developed and used for editing the staphylococcal phage genome
[35][106]. The CRISPR-Cas10 system provides more protection to the host bacteria by exhibiting high cleavage activity toward the staphylococcal phage.