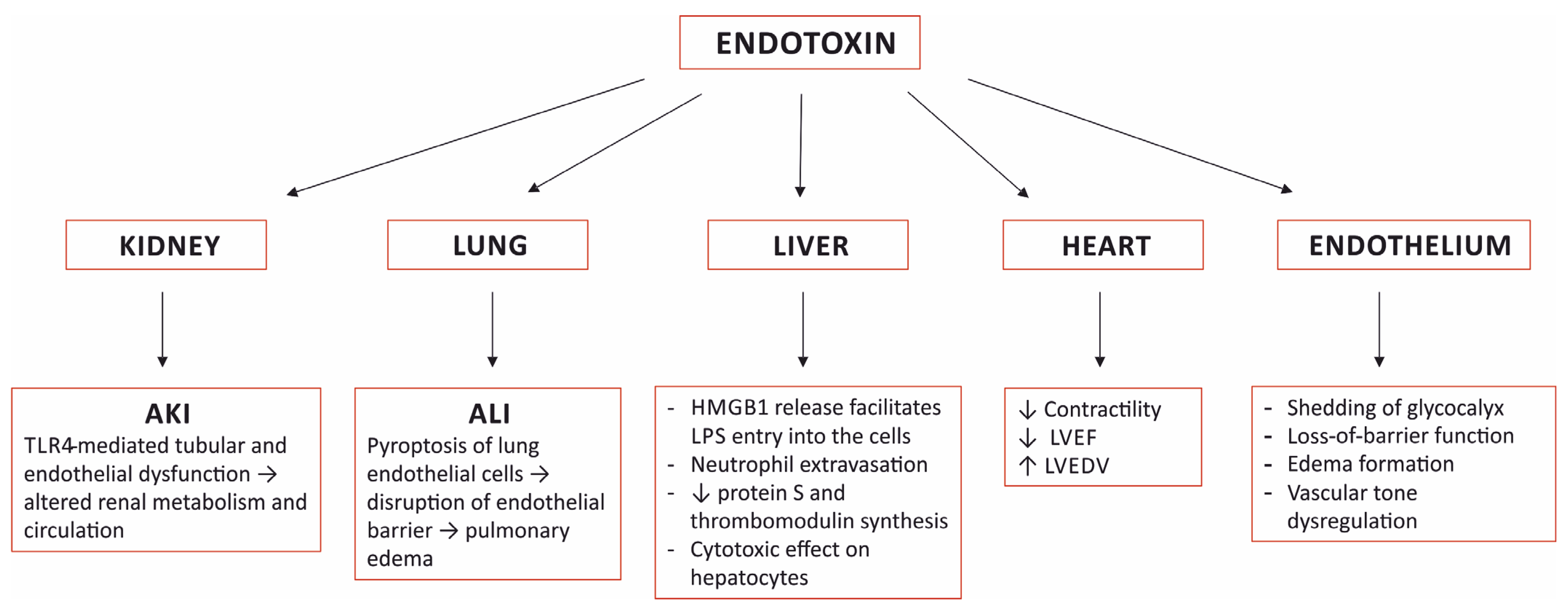
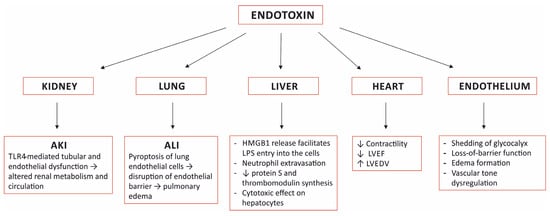
Lipopolysaccharide, the main component of the outer membrane of Gram-negative bacteria is a highly potent endotoxin responsible for organ dysfunction in sepsis. It is present in the blood stream not only in Gram-negative infections, but also in Gram-positive and fungal infections, presumably due to sepsis-related disruption of the intestinal barrier. Various pathways, both extra- and intracellular, are involved in sensing endotoxin and non-canonical activation of caspase-mediated pyroptosis is considered to have a major role in sepsis pathophysiology. Endotoxin induces specific pathological alterations in several organs, which contributes to poor outcomes. The adverse consequences of endotoxin in the circulation support the use of anti-endotoxin therapies, yet more than 30 years of experience with endotoxin adsorption therapies have not provided clear evidence in favor of this treatment modality. The results of small studies support timely endotoxin removal guided by measuring the levels of endotoxin; unfortunately, this has not been proven in large, randomized studies. The presence of endotoxemia can be demonstrated in the majority of patients with COVID-19, yet only case reports and case series describing the effects of endotoxin removal in these patients have been published to date. The place of blood purification therapies in the treatment of septic shock has not yet been determined.
- endotoxin
- septic shock
- blood purification
- bacterial translocation
1. Introduction
2. Lipopolysaccharide Sensing Pathways
2.1. Toll-like Receptor 4–Myeloid Differentiation Protein 2 (TLR4-MD-2) Pathway
2.2. Transient Receptor Potential (TRP) Ion Channels
2.3. Intracellular LPS Sensing
3. Organ Damage Caused by Sensing Endotoxin