Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jialei Ji and Version 3 by Dean Liu.
The SWEET (sugars will eventually be exported transporter) family was identified as a new class of sugar transporters that function as bidirectional uniporters/facilitators and facilitate the diffusion of sugars across cell membranes along a concentration gradient. SWEETs are found widely in plants and play central roles in many biochemical processes, including the phloem loading of sugar for long-distance transport, pollen nutrition, nectar secretion, seed fifilling, fruit development, plant–pathogen interactions and responses to abiotic stress.
- SWEET
- sugar transporter
- phloem loading
- plant–pathogen interaction
1. Structural Characteristics of the SWEETs
Before the discovery of SWEETs, the MSTs and SUTs that had been identified in plants were members of the major facilitator superfamily (MFS). The N-terminal and C-terminal ends of these proteins are both located on the intracellular side and typically contain 12 α-helical transmembrane domains (TMs). There is a large cytoplasmic loop located on the intracellular side in the middle of the MFS protein, which divides the protein into two domains [1][2][3][26,49,50]. The N-terminal and C-terminal domains each contain six TMs. The topological structures of these two domains are very similar and exist in a pseudo-quadratic axisymmetric manner. The six TMs that form each domain can be split into two groups of three TMs that symmetrically repeat units in an anti-parallel manner [1][2][3][26,49,50]. This unique folding method is designated MFS fold [4][51].
The plant SWEETs are members of the MtN3/saliva family (PF03083). Its N-terminus and C-terminus are located on the outside and inside of the cytoplasm, respectively. Plant SWEETs generally contain seven TMs (Figure 1a,b). The fourth TM is less conservative and primarily acts as a link. It divides the protein into two MtN3/saliva domains that each contain three TMs that form a “3-1-3” structure [5][10]. The three TMs of each MtN3/saliva domain are arranged in the form of “TM1-TM3-TM2” to form a triple-helix bundle (THB) (Figure 1a,b). It is apparent that the topological structure of SWEETs differs significantly from those of MSTs and SUTs. This difference could be an important reason why SWEET can transport sugar intracellularly to extracellularly. In addition, the SWEETs of prokaryotes only contain one MtN3/saliva domain composed of three TMs [6][52]. Therefore, they have been designated SemiSWEETs. It can be hypothesized that one MtN3/saliva domain in prokaryotes underwent replication or horizontal gene transfer, which is defined as the transmission of DNA between different genomes, during the process of evolution, which led to the production of SWEET proteins in eukaryotes that contain two MtN3/saliva domains.
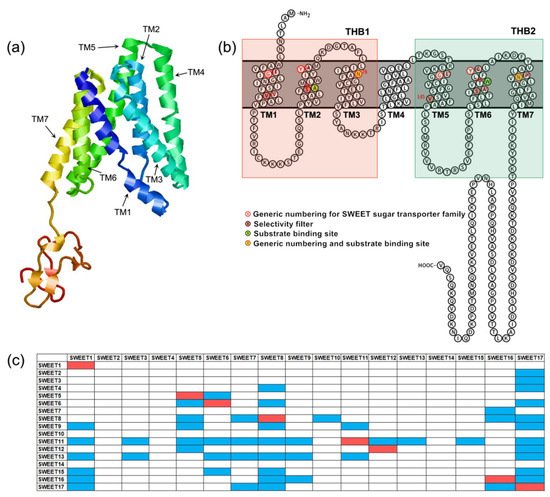
Figure 1. Structural characteristics of SWEETs. (a) The three-dimensional model of AtSWEET13 constructed with Phyre2. (b) Snake diagram of AtSWEET13 with key positions labeled. (c) AtSWEETs interaction matrix diagram [6][52]. The colored boxes represent interaction between two SWEETs. The red boxes indicate homopolymers, and the blue boxes indicate heteropolymers.
The results of truncation and complementation experiments demonstrated that SWEETs must undergo oligomerization to form homologous or heteromultimers to transport sugars [6][52]. The most likely scenario is that the eukaryotic SWEETs form dimers, while the prokaryotic SemiSWEETs form tetramers. A. thaliana SWEET proteins can form at least eight homopolymers and 47 heteropolymers [6][52] (Figure 1c). A high-resolution three-dimensional structural analysis of the bacterial SemiSWEET proteins proved that two SemiSWEET protein monomers form a basic translocation pore unit by forming a symmetrical homodimer [7][8][9][10][53,54,55,56]. In addition, the tryptophan residue from TM2 and the asparagine residue from TM3 are the key sites for the SemiSWEET protein to be able to transport sugars [7][8][9][10][53,54,55,56]. Rice OsSWEET2b was the first eukaryotic SWEET protein for which a three-dimensional structure was resolved [11][57]. It showed that a single OsSWEET2b protein monomer can form a basic translocation pore unit and that TM4 and THB1 are closely linked to constitute the N-terminal region. In addition, THB2 constitutes the C-terminal region. This explains why the truncation of AtSWEET1 protein into THB1 + TM4 and THB2 and their co-expression enables the transportation of glucose [6][52]. In contrast, THB1 and TM4 + THB2 that have been truncated and co-expressed cannot transport glucose [6][52]. Moreover, owing to the inconsistent tightness of TM4 with THB1 and THB2, THB1 and THB2 are structurally asymmetrical, which obviously differs from the symmetrical arrangement of the two THBs in the SemiSWEET homodimer of prokaryotes [11][57]. Cysteine residues from TM2, asparagine residues from TM3 and TM7 and phenylalanine residues from TM6 are the key sites for OsSWEET2 to transport glucose [11][57]. Recently, the crystal structure of A. thaliana AtSWEET13 with a resolution of 2.8-Å has been obtained. The researchers observed an inward-facing conformation of AtSWEET13 with the substrate analog 2′-deoxycytidine-5′-monophosphate bound to the central cavity [12][58]. There are 10 amino acid residues in the AtSWEET13 protein that play an important role in the recognition and binding of substrates. They are designated Ser20 from TM1 (Ser20TM1), Leu23TM1, Asn54TM2, Trp58TM2, Asn76TM3, Ser142TM5, Met145TM5, Asn176TM6, Trp180TM6 and Asn196TM7. Up to now, structural analysis has revealed that SemiSWEET or SWEET proteins have three conformations: outward open conformation, inward open conformation and occluded conformation. These results laid a structural foundation to elucidate the mechanism by which the SWEET protein binds to substrates and transports sugar. Based on this, a rocking-type motion theory was proposed [7][53]. In OsWEET2b, the proline residues on TM1, TM2, TM5 and TM6 may be the key factors that promote the transition between different conformations.
It is worth noting that there is a database designated dbSWEET (http://bioinfo.iitk.ac.in/bioinfo/dbsweet/Home, accessed on 7 December 2021) that contains more than 2000 SWEET members from prokaryotes and eukaryotes. This database helps researchers to more effectively analyze the biological functions of SWEET through targeted gene editing and simulation experiments.
2. Evolution of the SWEET Gene Family
Yuan and Wang [13][38] used the transporter classification database (TCDB; http://www.tcdb.org, accessed on 12 November 2021) to select SWEET proteins of different species for a phylogenetic analysis that revealed that the SWEET proteins in different species can be divided into three evolutionary clades. The SWEET proteins of monocots and dicots are members of Clade I, the SWEET proteins of metazoans and mammals are members of Clade II, and those of the bacteria and archaea are members of Clade III. Some members of the Caenorhabditis elegans MtN3/saliva family in metamorphosis are also included in Clade III. There is only one MtN3/saliva domain composed of three transmembrane helices in all the bacterial proteins in branch III. A phylogenetic analysis indicates that the widespread distribution of the MtN3/saliva SWEET protein may originate from the SemiSWEET protein of prokaryotes. Domain duplication occurred during the evolution of eukaryotes, resulting in a protein with seven transmembrane α-helices that contains two MtN3/saliva domains.
A further phylogenetic analysis of the SWEETs of 16 types of angiosperms revealed that the family was divided into four subfamilies that are designated Clade I~IV (Figure 2), which is consistent with the results of previous studies [14][39]. Utilizing A. thaliana as an example, the Clade I subfamily contains the three members AtSWEET1, -2 and -3; the Clade II subfamily has five members, including AtSWEET4, -5, -6, -7 and -8; the Clade III subfamily has seven members, including AtSWEET9, -10, -11, -12, -13, -14 and -15; and the Clade IV subfamily has the two members AtSWEET16 and -17. In addition, different subfamilies have selective preferences for monosaccharides or disaccharides. The Clade I and II subfamilies specifically transport hexose, the Clade III subfamily specifically transports sucrose, and the SWEET protein of the Clade IV subfamily is located on the vacuolar membrane and tends to transport fructose [15][59].
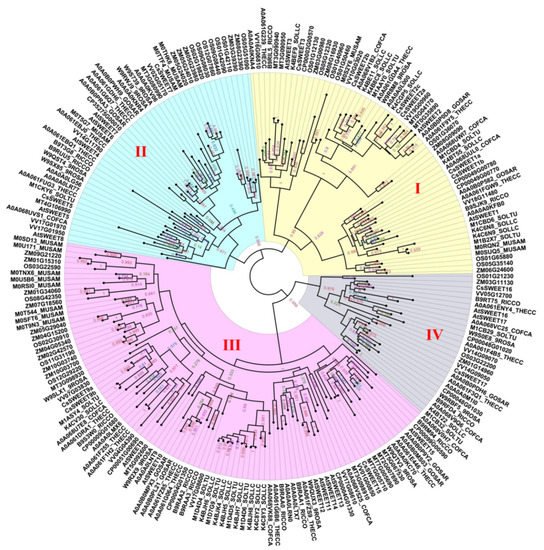
Figure 2. A phylogenetic tree of the SWEETs of 16 types of angiosperms. Accessions were identified from dbSWEET and UniProt. An alignment was conducted using MAFFT (v7.037). Phylogenetic trees were constructed using FastTree (http://www.microbesonline.org/fasttree/, accessed on 25 November 2021) based on the JTT + CAT model. The numerical values in the figure represent the reliability of the clades. The closer the value of the clade is to 1.0, the higher the confidence.
According to the existing evolutionary analysis, it is believed that the SWEET protein containing 7-TMs and having two MtN3/saliva domains in eukaryotes is produced by duplication of the MtN3/saliva domain containing 3-TMs in prokaryotes. This allows for more complex sucrose transport in eukaryotes [13][14][6][38,39,52]. More interestingly, the SWEET proteins of unicellular algae contain 7-TMs but have not yet formed a conserved THB unit, further speculating that multicellular plants (bryophytes and flowering plants) may acquire 3-TMs from symbiotic bacteria through horizontal gene transfer or possibly obtain 3-TMs through internal duplication.
3. SWEETs Participate in the Interaction between Host Plants and Pathogens
The SWEET gene family plays a key role in plant–pathogen interactions [16][17][18][19][20][91,92,93,94,95]. When bacterial or fungal pathogens invade plants, they secrete virulence proteins, described as transcription activator-like (TAL) effectors that can bind to the promoters of specific SWEET genes and activate their expression, which helps the pathogens to obtain sugars from plants for their growth and reproduction (Figure 34). Rice OsSWEET11-15 has been proven to provide nutrition for rice bacterial blight (Xanthomonas oryzae pv. oryzae) [21][18][20][22][23][24][25][26][27][68,93,95,96,97,98,99,100,101]. X. pv. oryzae infects rice and secretes specific TAL effectors, which combine with the specific elements (effector-binding elements, EBEs) of the promoter of the target OsSWEET gene to induce the up-regulated expression of the gene. This leads to the outflow of more carbohydrates so that the pathogenic bacteria can obtain nutrition and multiply. This increases the susceptibility of the plant to disease. TAL effectors that can induce the expression of OsSWEET11-15 have been identified. Among them, four TAL effectors have been identified that can induce the expression of OsSWEET14 [18][20][22][23][24][93,95,96,97,98]. After suppressing OsSWEET11, the rice plants were resistant to sheath blight disease caused by the fungus Rhizoctonia solani [28][102].
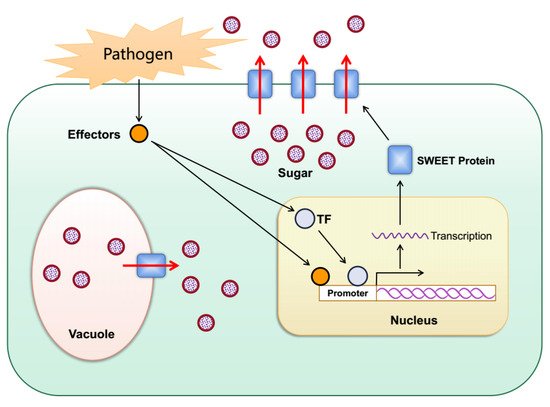
Figure 34. Schematic diagram for the role of plant SWEETs in pathogen nutrition. When bacterial or fungal pathogens invade plants, they secrete TAL effectors into host plant cells. The TAL effectors induce the expression of plant SWEETs either directly or indirectly through activation of transcription factors, resulting in the outflow of sugar into the apoplast as a source of nutrition for the pathogens.
The phenomenon of pathogen-induced SWEET gene expression is also found in other plant species. Cassava (Manihot esculenta) MeSWEET10a can be induced by the TAL20 effector of X. axonopodis pv. Manihotis [29][103]. Orange CsSWEET1 can be induced by the PthA4 and PthAw effectors of bacterial citrus canker (X. citri ssp. citri) [30][104]. Grape VvSWEET4 and VvSWEET7 can interact with gray mold (Botrytis cinerea) [31][32][40,105]. The level of expression of VvSWEET4 and VvSWEET7 was up-regulated after B. cinerea infected grapes [33][18][35,93]. The atsweet4 mutant is not sensitive to B. cinerea [31][40]. The level of expression of AtSWEET2 increases sharply after the oomycete Pythium irregulare infects the roots of A. thaliana, while the atsweet2 mutant is not sensitive to Pythium [34][45]. This suggests that the location of AtSWEET2 in the vacuole membrane enables it to provide glucose that Pythium can use to grow and reproduce. The accumulation of sugar in the leaves of A. thaliana sweet11;sweet12 double mutants can trigger a defense pathway against the fungal pathogen Colletotrichum higginsianum mediated by salicylic acid [35][106]. The AtSWEET genes in A. thaliana are also induced by bacterial speck disease (Pseudomonas syringae pv. tomato DC3000), powdery mildew (Golovinomyces cichoracearum) and clubroot (Plasmodiophora brassicae) [33][36][35,107]. The level of expression of the SWEET gene (UPA16) in pepper (Capsicum annuum) is induced by black rot (X. campestris pv. vesicatoria) [37][108]. The level of expression of the SWEET gene in wheat is induced by the rust fungus Puccinia striiformis [38][109]. GhSWEET10 in cotton (Gossypium hirsutus) can be induced by Avrb6, a TAL effector that determines the pathogenicity of X. citri subsp. malvacearum (Xcm) [39][110]. Silencing GhSWEET10 reduces the susceptibility of cotton to Xcm [39][110]. The infection of P. brassicae on B. rapa can induce the translocation of sugar between the tissues that produce sugar and that of the susceptible clubbed tissue. BrSWEETs participate in this process [40][111]. Unlike the above cases, IbSWEET10 in sweet potato (Ipomoea batatas) plays a positive role in resistance to the fungus Fusarium oxysporum. The overexpression of IbSWEET10 can increase the resistance of sweet potato to F. oxysporum by reducing the sugar content of sweet potato [41][112].
The results described above indicate that the SWEET gene family participates in host–pathogen interactions, but only a few SWEET genes and mechanisms of their interaction with pathogens have been explained. Elucidating these mechanisms will help researchers to modify the binding sites of pathogens through molecular biological methods to obtain crops that are resistant to pathogens. A number of bioinformatics tools, such as PrediTALE, Storyteller, Talvez, TALgetter and Target Finder, can be used to identify and predict EBEs for almost all known TAL effectors [42][113]. Resistant plants can be obtained by modifying EBEs using gene-editing technology. For example, The CRISPR-Cas9 system was used to edit the EBEs in the promoter regions of SWEET11, SWEET13 and SWEET14, resulting in rice plants that were resistant to the main pathogens that cause rice blight [43][114]. In this regard, reswearchers systematically summarize a strategy to obtain disease-resistant plants by modifying SWEET genes (Figure 45). Moreover, some studies have shown that SWEET family genes not only provide carbohydrates for pathogenic bacteria but also provide nutrients for beneficial microorganisms. For example, alfalfa MtSWEET11 can be induced by rhizobia to provide carbohydrates for the formation of symbiotic nodules [44][62].
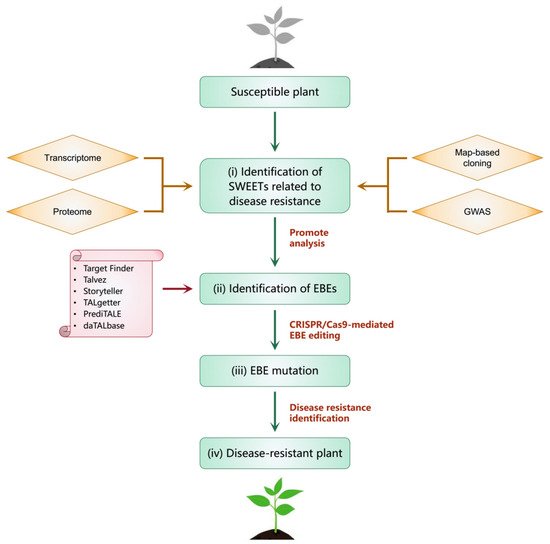
Figure 45. The strategy to obtain disease-resistant plants by modifying SWEET genes. It mainly includes the following steps (i–iv). (i) Identify SWEETs related to disease resistance through methods such as map-based cloning, genome-wide association analysis (GWAS), transcriptome and proteome. (ii) Identify and predict EBEs through tools such as PrediTALE, Storyteller, Talvez, TALgetter and Target Finder. (iii) Modify EBEs using gene-editing technology such as CRISPR-Cas9 system. (iv) Disease resistance test and obtain disease-resistant plants.