The family of NF-κB is composed of five members of proteins linking to DNA, (RelA, RelB, RelC, NFκ-B1, and NFκ-B2) that trigger a set of inflammatory downstream effectors after nuclear translocation, involved in a broad range of biological processes. Either a canonical or a non-canonical pathway can be responsible for their activation; the canonical pathway mediates inflammatory responses and leads to a rapid but transient NF-κB activation, while the non-canonical signalling is a slow, long-lasting pathway.
- NF-kB
- Sjogren's syndrome
- inflammation
1. Introduction
Typical inducers of the non-canonical NF-κB pathway are ligands of a subset of the tumour necrosis factor receptors (TNFR) superfamily involved in the differentiation of the immune system, as well as in secondary lymphoid organogenesis [1]. The NF-κB family members show homology through a 300 amino acid N-terminal DNA binding/dimerization domain, named the Rel homology domain (RHD). The RHD is a complex system where family members can constitute homodimers and heterodimers, which are normally kept inactive in the cytoplasm through interaction with inhibitory proteins of the IκB family (IκBs) [2]. The common regulatory step in both the canonical and non-canonical cascades is the activation of an IκB kinase (IKK) complex consisting of catalytic kinase subunits (IKKα and/or IKKβ) and the regulatory non-enzymatic scaffold protein NF-κB essential modulator (NEMO), also known as IKKγ [2]. NF-κB dimers are activated by IKK-mediated phosphorylation of IκBs, which triggers proteasomal IκBs degradation, liberating the dimers of NF-κB from the NF-κB-IκBs complex, that subsequently translocate to the nucleus, linking to κB enhancer elements of target genes [2] (Figure 1). Among the IκBs, the best characterized is IκBα, which requires degradation of the activity of IKK kinase. IκBα functions as a negative feedback loop to sequester NF-κB subunits, because NF-κB activation induces the expression of the IκBα gene, which terminates signalling unless a persistent activation signal is present [2].
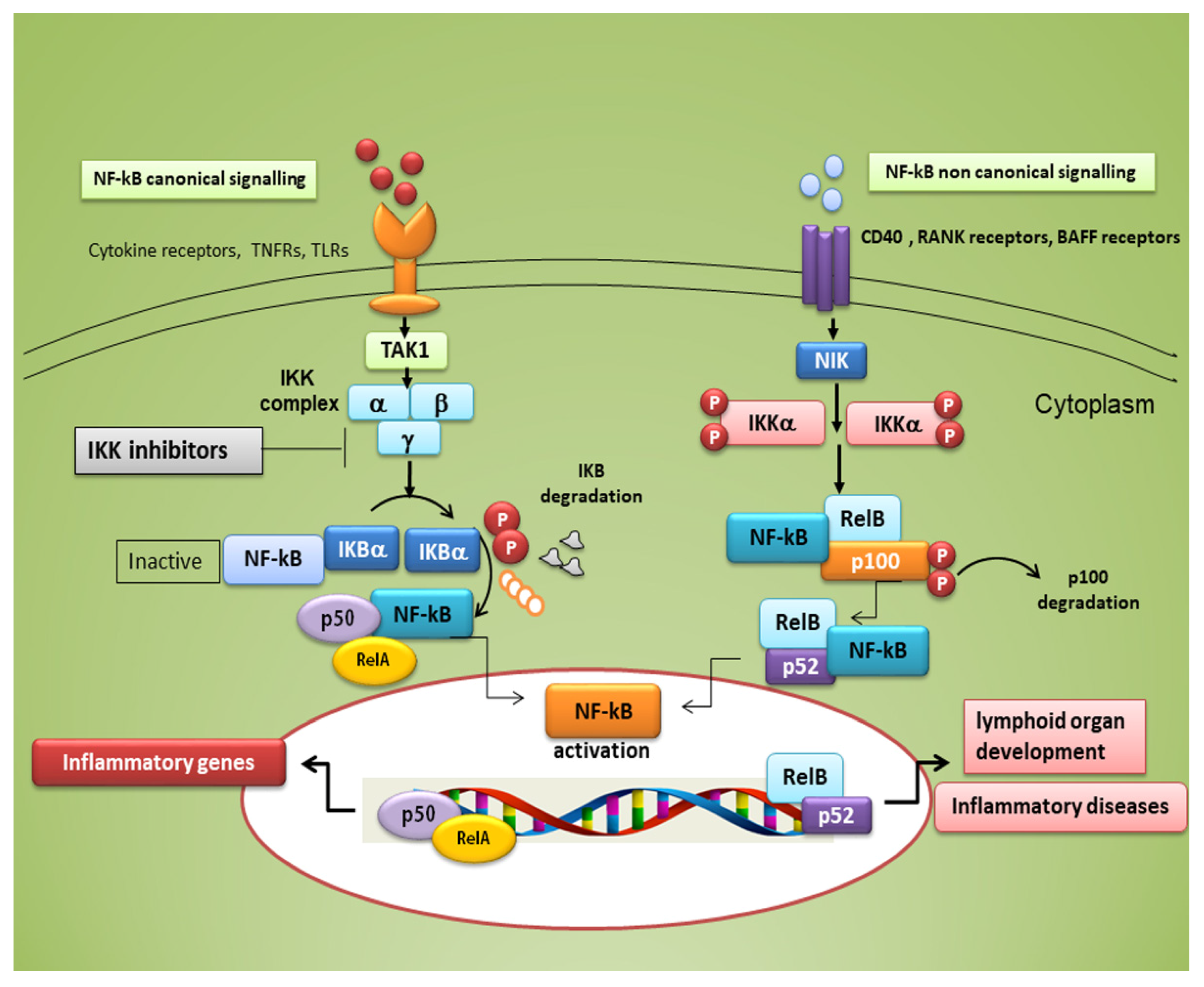
Figure 1. Canonical and non-canonical nuclear factor κ (kappa)-light-chain-enhancer of activated B cells (NF-κB) signalling pathways. The canonical NF-κB pathway starts with the activation of innate and adaptive immune receptors in response to various ligand molecules, which transfers the signal across the cell membrane causing the activation of the trimeric IκB kinase (IKK) complex, composed of catalytic (IKKα and IKKβ) and regulatory (IKKγ) subunits. The IKK complex phosphorylates IκBα and phosphorylated IκB undergo ubiquitylation and proteasomal degradation, allowing nuclear translocation of the RelA/p50 dimer of the NF-κB heterodimer. The non-canonical NF-κB pathway selectively responds to a subset of TNFR members that induce the activation of the NF-κB-inducing kinase (NIK). NIK phosphorylates and activates IKKα, which in turn phosphorylates carboxy-terminal serine residues of p100, triggering selective degradation of the C-terminal IκB-like structure of p100, and mediates the persistent activation of the RelB/p52 complex.
NF-κB is activated in every cell type and has a central role in inflammation. It is, therefore, fundamentally implicated in the molecular pathways that induce the transcription of pro-inflammatory genes [3][4][5][6][7][8]. NF-κB is highly activated in various inflammatory disorders and triggers the transcription of chemokines, cytokines, pro-inflammatory enzymes, adhesion proteins, and other factors to modulate the inflammatory response, such as metalloproteinases, Cox-2, and inducible nitric oxide synthase [9][10][11], although the mechanism is still unclear (a schematic representation of NF-κB activation is reported in Figure 2). In RA, NF-κB is highly expressed in the inflamed synovial stratum [12][13], where it enhances the engagement of inflammatory cells and pro-inflammatory cytokines production such as IL-1, IL-6, IL-8, and TNF-α [12][13]. Interestingly, recent evidence has shown that alterations in the modulation of NF-κB-dependent gene expression lead to a variety of other inflammatory and autoimmune disorders, neurological conditions and cancers [7][8]. In pSS, a correlation between NF-κB signalling and chronic inflammation has been demonstrated by various reports; nuclear translocation of NF-κB into focal infiltrated lymphocytes and into the acinar epithelium surrounding the infiltrates from SGs of patients with pSS was detected, while distal normal acini and ductal structures showed no nuclear translocation [14]. In addition, the non-canonical NF-κB p65 nuclear translocation has been induced in pSS SGEC by a range of molecular agents such as epidermal growth receptor (EGFR) and B-cell activator CD40 [15][16]. Furthermore, a downregulated gene and protein expression of IκBα was detected in pSS monocytes, contributing to an enhanced NF-κB activity. Finally, pSS autoantibodies can trigger the NF-κB signalling pathway, thus, contributing to exacerbate the inflammatory condition [17].
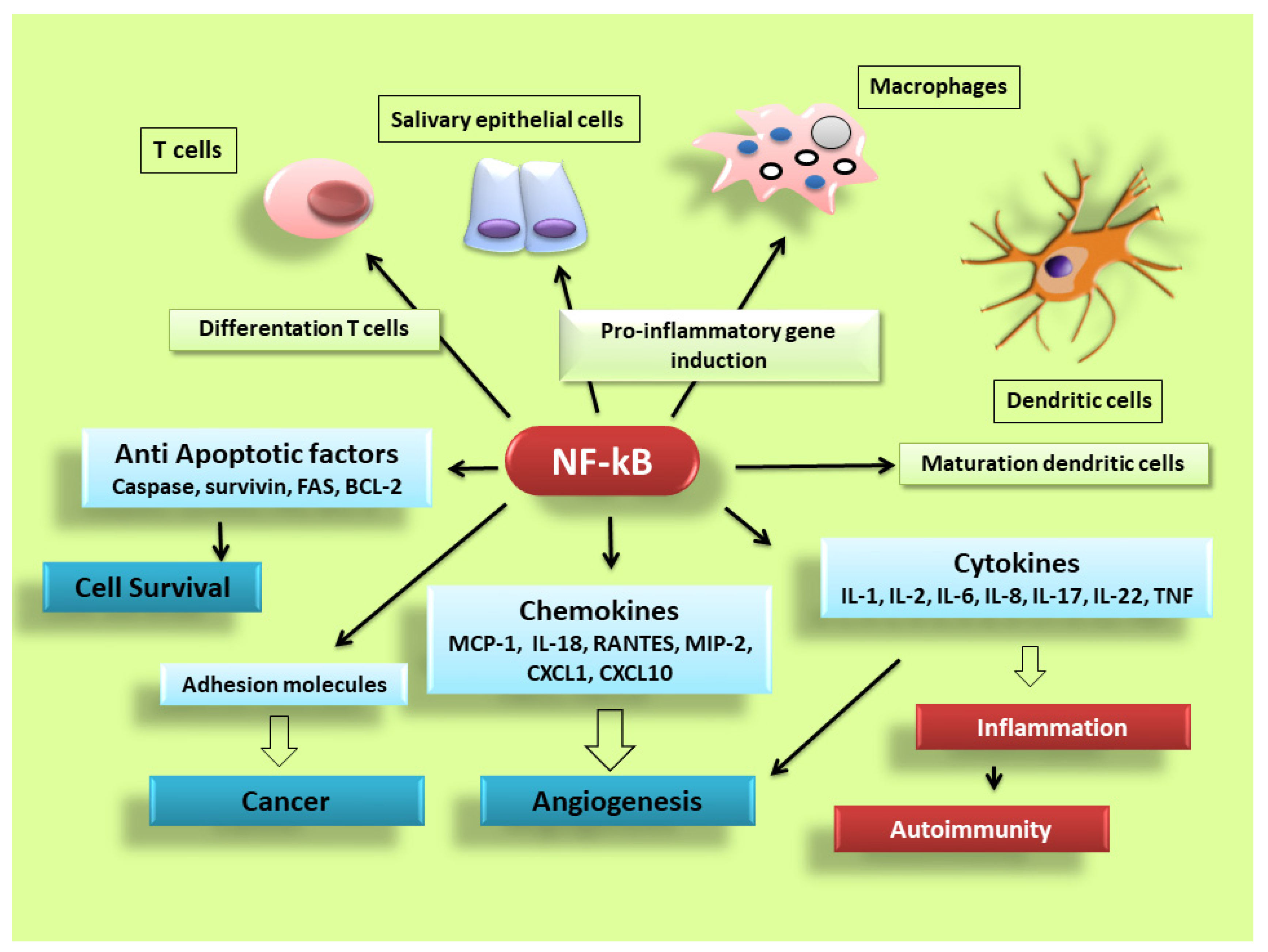
Figure 2. NF-κB signalling pathway dictates the inflammatory responses. After its activation, it can induce the transcription of a large number of genes including pro-inflammatory cytokines, chemokines, adhesion molecules, cell cycle regulatory molecules, anti-apoptotic proteins and angiogenic factors, and thereby regulate cell proliferation, apoptosis, morphogenesis, differentiation, angiogenesis, and inflammation. The regulation of inflammation, cell proliferation and apoptosis is central to the understanding of many diseases, such as autoimmune diseases and cancer.
2. Small-Molecule Inhibitors of NF-κB in Sjögren's Syndrome
Sjögren's syndrome (SS) is a systemic autoimmune inflammatory disease with a poorly defined aetiology, which targets exocrine glands (particularly salivary and lachrymal glands), affecting the secretory function. Patients suffering from SS exhibit persistent xerostomia and keratoconjunctivitis sicca. It is now widely acknowledged that a chronic grade of inflammation plays a central role in the initiation, progression, and development of SS. Consistent with its key role in organizing inflammatory responses, numerous recent studies have shown involvement of the transcription factor nuclear factor κ (kappa)-light-chain-enhancer of activated B cells (NF-κB) in the development of this disease. Therefore, chronic inflammation is considered as a critical factor in the disease aetiology, offering hope for the development of new drugs for treatment.
The modulation of the NF-κB pathways has frequently been described as “pro-inflammatory”, largely due to the key role of NF-κB in the pro-inflammatory genes expression including cytokines, chemokines, and adhesion molecules [18][19]. Many findings demonstrated that epithelial cells in the glandular sites of patients affected by pSS are able to release factors that address the chemoattraction of lymphocytes and promote chronic inflammatory responses [20][21][22][23][5][6]. NF-κB pathway modulation was therefore investigated in pSS, highlighting a role in regulating the production of pro-inflammatory cytokines, leukocyte enrolment, or cell survival [3][4][5][6][24]. In pSS, the NF-κB activation cascade can be modulated at different levels [1]. Considering the correlation with the biopsy focus score, grade of infiltration and evaluated disease activity, phosphorylated IKKε, responsible for the degradation of IkB proteins, were significantly and positively correlated with NF-κB levels in pSS [25]. The levels of B-Cell Activating Factor (BAFF) and those of numerous pro-inflammatory cytokines, all regulated by NF-κB signalling, are augmented in pSS [26]. Nucleotide polymorphisms in NF-κB pathway genes have been linked with pSS [27], and a specific mutation in the Ikα-826T, one of the promoters of a member of the inhibitory IkB complex, was associated with susceptibility of pSS [28][29]. Numerous small molecule inhibitors of the NF-κB signalling pathways are currently commercially available for use, and NF-κB modulators are under study in clinical trials for pSS treatment [30][5][1][31][32][33][34][35][36]. Many preclinical studies have already analysed the role of NF-κB signalling in the glandular tissue in pSS. The pSS SGECs have been recognized to have an active NF-κB pathway. The phosphorylated forms of IKKε, IκBα, and NF-κB were expressed in the ductal cells in minor SGs derived from pSS patients [30]. By stimulating the Toll-Like Receptor 2 (TLR2) in SGECs, IL-2 production was induced through the NF-κB cascade in pSS SGECs [31][32][33]. SGECs treated with the anti-Ro/SSA autoantibodies isolated from pSS patients showed a progressive increase in constitutive NF-κB activation, and transfection of SGECs with IκBα in SGECs treated with anti-Ro/SSA led to a remarkable production of pro-inflammatory cytokines and an enhanced apoptosis [5]. Furthermore, recent findings showed that gene silencing of the natural NF-κB inhibitor TNF Alpha Induced Protein 3 (TNFAIP3) in keratin-14-positive epithelial cells, promoting the activation of the constitutive NF-κB cascade, induces the initial phases of pSS, leading to a reduced production of saliva and lymphocyte invasion in the SGs [34]. This effect is likely related to the calcium pathway in the acinar cells, since calcium signalling has an important role in NF-κB pathway activity [35][36]. A list of NF-κB small molecule inhibitors tested in pSS is reported in Table 1.
Table 1. List of NF-κB small molecules inhibitors tested or identified in primary SS (pSS).
| Small Inhibitors of NF-kB in pSS | Study | References |
|---|
| Iguratimod | Clinical study | [1] |
| Syk-inihibitor-Gs-9876 | Clinical study | [1] |
| IKKε | Pre-clinical study | [25][1] |
| IκBα | Pre-clinical study | [30][6][28][29] |
| anti-Ro/SSA autoantibodies | Pre-clinical study | [37][22][23][5] |
| TNFAIP3 | Pre-clinical study | [3] |
| Calcium mobilization | Pre-clinical study | [35][36] |
3. Conclusion
This entry presents that NF-κB has a crucial role in the pathogenesis of pSS, since it promotes chronic inflammation. NF-κB, through intrinsic SGEC activation, regulates sophisticated feedback circuits in pSS that comprise all elements of the cellular immune response. Since NF-κB and members of its signalling pathways regulate cellular activity from DNA transcription to translation into proteins, efficient and properly controlled NF-κB signalling is important during physiological immune homeostasis. In fact, the integrity of the signal triggered by NF-κB is essential in preventing the onset of pSS autoimmune disease. It is noteworthy that the significance of NF-κB activation in pSS suggests that inhibition of this signalling pathway could provide novel strategies for the prevention and treatment of the SGs dysfunction characterizing pSS. New future perspectives suggest, for example, the use of IKKε inhibitors for the treatment of pSS, repressing downstream NF-κB signalling activation. Furthermore, great attention is now being paid to the modulation of NF-κB activity and expression of NF-κB target genes through an IκBα-mediated negative feedback mechanism. Hopefully, as our understanding of the regulation of the NF-κB pathways increases, insights into a better design of drugs that can effectively target NF-κB for the prevention and treatment of pSS may be gained.
References
- Pringle, S.; Wang, X.; Bootsma, H.; Spijkervet, F.K.L.; Vissink, A.; Kroese, F.G.M. Small-molecule inhibitors and the salivary gland epithelium in Sjögren’s syndrome. Expert Opin. Investig. Drugs. 2019, 28, 605–616.
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234.
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Ingravallo, G.; Maiorano, E.; D’Amore, M. A failure of TNFAIP3 negative regulation maintains sustained NF-κB activation in Sjögren’s syndrome. Histochem. Cell Biol. 2011, 135, 615–625.
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; D’Amore, M.; Frassanito, M.A.; Ribatti, D. Sjögren’s syndrome pathological neovascularization is regulated by VEGF-A-stimulated TACE-dependent crosstalk between VEGFR2 and NF-κB. Genes Immun. 2012, 13, 411–420.
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M. Sjögren’s syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF-κB. Lab. Investig. 2012, 92, 615–624.
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M. Altered IκBα expression promotes NF-κB activation in monocytes from primary Sjögren’s syndrome patients. Pathology 2012, 44, 557–561.
- Christman, J.W.; Sadikot, R.T.; Blackwell, T.S. The role of nuclear factor-kappa B in pulmonary diseases. Chest 2000, 117, 1482–1487.
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-kB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 2001, 1, 287–296.
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734.
- Kaltschmidt, B.; Widera, D.; Kaltschmidt, C. Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 2005, 1745, 287–299.
- Ledoux, A.C.; Perkins, N.D. NF-kappaB and the cell cycle. Biochem. Soc. Trans. 2014, 42, 76–81.
- Han, Z.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. AP-1 and NF-kappa B regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 1998, 28, 197–208.
- Makarov, S.S. NF-kappa B in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001, 3, 200–206.
- Wang, X.; Shaalan, A.; Liefers, S.; Coudenys, J.; Elewaut, D.; Proctor, G.B.; Bootsma, H.; Kroese, F.G.M.; Pringle, S. Dysregulation of NF-kB in glandular epithelial cells results in Sjögren’s-like features. PLoS ONE 2018, 13, e0200212.
- Nakamura, H.; Kawakami, A.; Ida, H.; Koji, T.; Eguchi, K. EGF activates PI3K-Akt and NF-κB via distinct pathways in salivary epithelial cells in Sjögren’s syndrome. Rheumatol. Int. 2007, 28, 127–136.
- Ping, L.; Ogawa, N.; Zhang, Y.; Sugai, S.; Masaki, Y.; Xiao, W. p38 mitogen-activated protein kinase and nuclear factor-kappaB facilitate CD40-mediated salivary epithelial cell death. J. Rheumatol. 2012, 39, 1256–1264.
- Sisto, M.; Lisi, S.; Castellana, D.; Scagliusi, P.; D’Amore, M.; Caprio, S.; Scagliusi, A.; Acquafredda, A.A.; Panaro, M.A.; Mitolo, V. Autoantibodies from Sjögren’s syndrome induce activation of both the intrinsic and extrinsic apoptotic pathways in human salivary gland cell line A-253. J. Autoimmun. 2006, 27, 38–49.
- Barnes, P.; Karin, M. Nuclear Factor-κB: A Pivotal Transcription Factor in Chronic Inflammatory Diseases. N. Engl. J. Med. 1997, 336, 1066–1071.
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11.
- Moutsopoulos, H.M. Sjögren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165.
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Caprio, S.; Mitolo, V.; D’Amore, M. TNF blocker drugs modulate human TNF-α-converting enzyme pro-domain shedding induced by autoantibodies. Immunobiology 2010, 215, 874–883.
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Ingravallo, G.; Mitolo, V.; D’Amore, M. Expression of pro-inflammatory TACE-TNF-α-amphiregulin axis in Sjögren’s syndrome salivary glands. Histochem. Cell Biol. 2010, 134, 345–353.
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; Cucci, L.; Frassanito, M.A.; Mitolo, V.; D’Amore, M. Pro-inflammatory role of Anti-Ro/SSA autoantibodies through the activation of Furin-TACE-amphiregulin axis. J. Autoimmun. 2010, 35, 160–170.
- Dale, E.; Davis, M.; Faustman, D.L. A role for transcription factor NF-κB in autoimmunity: Possible interactions of genes, sex, and the immune response. Adv. Physiol. Educ. 2006, 30, 152–158.
- Chen, W.; Lin, J.; Cao, H.; Xu, D.; Xu, B.; Xu, L.; Yue, L.; Sun, C.; Wu, G.; Qian, W. Local and Systemic IKKε and NF-κB Signaling Associated with Sjögren’s Syndrome Immunopathogenesis. J. Immunol. Res. 2015, 2015, 534648.
- Thompson, N.; Isenberg, D.A.; Jury, E.C.; Ciurtin, C. Exploring BAFF: Its expression, receptors and contribution to the immunopathogenesis of Sjögren’s syndrome. Rheumatology 2016, 55, 1548–1555.
- Nordmark, G.; Wang, C.; Vasaitis, L.; Eriksson, P.; Theander, E.; Kvarnstrom, M.; Forsblad-d’Elia, H.; Jazebi, H.; Sjowall, C.; Reksten, T.R.; et al. Association of Genes in the NF-kappaB Pathway with Antibody-Positive Primary Sjogren’s Syndrome. Scand. J. Immunol. 2013, 78, 447–454.
- Ou, T.T.; Lin, C.H.; Lin, Y.C.; Li, R.N.; Tsai, W.C.; Liu, H.W.; Yen, J.H. IkappaBalpha promoter polymorphisms in patients with primary Sjögren’s syndrome. J. Clin. Immunol. 2008, 28, 440–444.
- Peng, B.; Ling, J.; Lee, A.J.; Wang, Z.; Chang, Z.; Jin, W.; Kang, Y.; Zhang, R.; Shim, D.; Wang, H.; et al. Defective feedback regulation of NF-kappaB underlies Sjogren’s syndrome in mice with mutated kappaB enhancers of the IkappaBalpha promoter. Proc. Natl. Acad. Sci. USA 2010, 107, 15193–15198.
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Ingravallo, G.; De Lucro, R.; D’Amore, M. Salivary gland expression level of IκBα regulatory protein in Sjögren’s syndrome. J. Mol. Histol. 2013, 44, 447–454.
- Kwok, S.K.; Cho, M.L.; Her, Y.M.; Oh, H.J.; Park, M.K.; Lee, S.Y.; Woo, Y.J.; Ju, J.H.; Park, K.S.; Kim, H.Y.; et al. TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren’s syndrome. Arthritis Res. Ther. 2012, 14, R64.
- Sisto, M.; Lorusso, L.; Lisi, S. Interleukin-15 as a potential new target in Sjögren’s syndrome-associated inflammation. Pathology 2016, 48, 602–607.
- Sisto, M.; Lorusso, L.; Lisi, S. TLR2 signals via NF-κB to drive IL-15 production in salivary gland epithelial cells derived from patients with primary Sjögren’s syndrome. Clin. Exp. Med. 2017, 17, 341–350.
- Das, T.; Chen, Z.; Hendriks, R.W.; Kool, M. A20/Tumor Necrosis Factor α-Induced Protein 3 in Immune Cells Controls Development of Autoinflammation and Autoimmunity: Lessons from Mouse Models. Front. Immunol. 2018, 9, 104.
- Dawson, L.J.; Field, E.A.; Harmer, A.R.; Smith, P.M. Acetylcholine-evoked calcium mobilization and ion channel activation in human labial gland acinar cells from patients with primary Sjogren’s syndrome. Clin. Exp. Immunol. 2001, 124, 480–485.
- Lilienbaum, A.; Israël, A. From calcium to NF-kappa B signaling pathways in neurons. Mol. Cell. Biol. 2003, 23, 2680–2698.
- Sisto, M.; Lisi, S.; Lofrumento, D.; D’Amore, M.; Scagliusi, P.; Mitolo, V. Autoantibodies from Sjögren’s syndrome trigger apoptosis in salivary gland cell line. Ann. N. Y. Acad. Sci. 2007, 1108, 418–425.