Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Hueng-Chuen Fan and Version 2 by Catherine Yang.
Glioblastoma multiforme (GBM) is a type of brain tumor that is notorious for its aggressiveness and invasiveness, and the complete removal of GBM is still not possible, even with advanced diagnostic strategies and extensive therapeutic plans. Its dismal prognosis and short survival time after diagnosis make it a crucial public health issue.
- glioblastoma multiforme (GBM)
1. Isocitrate Dehydrogenase (IDH)
To elucidate genetic alterations in GBM patients with varying prognoses or responses to specific targeted therapies and to identify subgroups of GBM patients for a better histopathological classification, an integrated genomic analysis was used to identify mutations inisocitrate dehydrogenase 1(IDH1) in 12% of patients with GBM [1][46]. It was subsequently reported that GBMs without IDH1 mutations often have mutations of isocitrate dehydrogenase 2 (IDH2) [2][47]. The structures of IDH1 (located on chromosome 2q33.3) and IDH2 (located on chromosome 15q26.1) are homodimeric and share similar sequences and an almost identical protein structure [3][48]. IDH1 and IDH2 encode two separate, different isocitrate dehydrogenase enzymes, which are nicotinamide adenine dinucleotide phosphate (NADP+)-dependent, catalyze oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG), and reduce oxidized nicotinamide adenine dinucleotide (NAD+) or NADP+ to NADH or NADPH. IDH1, localizing in the cytoplasm and peroxisomes, involves cellular metabolism and protection from reactive oxygen species and radiation [4][5][49,50]. IDH2, localizing in the mitochondria, is associated with the regulation of the tricarboxylic acid cycle and protection from oxidative stress [5][50]. DH1 and IDH2 mutations are mono-allelic, somatic, and missense changes. Mutations in IDH1 commonly affect R132, which is the binding site for isocitrate [1][46]. Mutations in IDH2 only affect R172 and R140 [2][6][47,51]. Mutant IDH possesses catalytic activity to convert α-KG to 2-hydroxyglutarate (2-HG) [7][52]. Excessive 2-HG is a metabolic hallmark of certain subtypes of gliomas [8][53]. Both mutated IDH1 and IDH2 are common in adult gliomas (WHO grades II and III) and secondary GBM (WHO grade IV). Mutated IDH1 and IDH2 are very rare in childhood GBM [9][54], suggesting gliomas with mutated IDH are clinically and genetically different from those with wild-type (WT) IDH genes.
2. Co-Deletion at Chromosome Regions 1p/19q
The complete deletion of chromosomes 1p and 19q is common in oligodendrogliomas and occur in 50–70% of both low-grade and anaplastic tumors [10][11][12][13][55,56,57,58]. These findings suggest that chromosomes 1p and 19q may contain tumor suppressor genes, including the far upstream element binding protein 1 (FUBP1) on chromosome 1p and the capicua transcriptional repressor (CIC) on chromosome 19q [14][15][16][59,60,61]. The FUBP1 expresses a single-stranded DNA-binding protein that can bind to several DNA regions, which harbor the far upstream element (FUSE) that is localized in the upstream of c-Myc. One function of FUSE is to regulate c-Myc in undifferentiated cells [17][62]. CIC, a tissue-specific transcriptional repressor, is expressed in the developing CNS and its dysfunction is associated with spinocerebellar ataxia type 1. This CIC-DNA interaction can be inhibited through the activation of the receptor tyrosine kinase (RTK) core signaling molecule mitogen-activated protein kinase (MAPK), which then allows for the transcription of CIC targets through this RTK-MAPK signaling axis. CIC alterations are common in specific cancer types (e.g., oligodendroglioma and Ewing-like sarcomas) [18][63]. Two clinical trials have clarified associations between combined 1p/19q co-deletion and an improved chemotherapeutic response and prognosis in oligodendrogliomas [19][20][64,65]. However, partial 1p or 19q deletion is more common in astrocytic tumors and secondary GBM but rare in primary GBM [21][22][23][66,67,68].
3. Mutations of H3F3A
Inside the nucleus, DNA, RNA and proteins form chromatin, which packs the DNA to a smaller volume and prevents the long DNA strands from being tangled. The structure of the chromatin is like “beads on a string”. The nucleosome is the “bead”, which is a basic unit of chromatin. Histones, nuclear proteins, can store DNA, modulate chromatin structure, impact gene expression, and regulate almost all DNA metabolic processes through post-translational modification, which includes methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation [24][69]. Each nucleosome is composed of a histone octamer, which is composed of two copies each of the core histone H2A, H2B, H3, and H4. H1, which does not contribute to the nucleosome bead, binds to the linker DNA region between nucleosomes, helping pack the chromatin into higher order structures. A chain of nucleosomes is compacted to form a chromosome [25][70]. Histone H3s, which have several variants, include H3.1, H3.2, and H3.3. H3.1 is encoded by HIST1H3A-J.H3.2 is encoded by HIST2H3A, HIST2H3C, and HIST2H3D. H3.3 is encoded by H3F3A and H3F3B [26][71]. Numerous H3 lysine residues can be post-translationally modified, including acetylated at lysines 9, 14, 18, 23, and 56; methylated at arginine 2 and lysines 4, 9, 27, 36, and 79, and phosphorylated at ser10, ser28, Thr3, and Thr11 [27][72]. Mutation of H3F3A, including K27M substitution, in which a lysine residue on the histone H3 tail is substituted for a methionine, were discovered in diffused intrinsic pontine glioma(DIPG) [28][73]. As the location of the H3F3A at lysine 27 is at or near critical regulatory histone residues, therefore, the alternations of mutant K27M on genes, which should be silent, produce widespread aberrant DNA methylation and deregulation of gene expression, impede physiological differentiation and to drive cell transformation [29][74]. Furthermore, in the WHO CNS tumor classification 2016, the principle of an integrated diagnosis was introduced with the combination of histological and molecular features, exemplified in the novel entity “diffuse midline glioma, H3K27M- mutant” [30][5]. The mutation in HIST1H3B-C, have been detected in approximately 10% of DIPG [31][75]. Another mutation of H3F3A, encoding a glycine 34 to arginine or valine (G34R/V) substitution, is reported in a smaller portion of pediatric and young adult high-grade astrocytoma [9][54]. H3F3G34 mutations may drive pediatric GBM through mismatch repair (MMR) deficiency [32][76] and upregulation of v-myc myelocytomatosis viral related oncogene, neuroblastoma derived (avian) (MYCN) [33][77].
4. Telomeres and Telomerase
A telomere is a capping structure located at the end of a linear chromosome. As most prokaryotes have circular chromosomes, they do not have telomeres; on the other hand, telomeres in vertebrates consist of a region of repeats of the six-nucleotide sequence TTAGGG at the ends of chromosomes, which themselves carry the complementary DNA strand sequence, AATCCC. In humans, the TTAGGG sequence repeats in tandem approximately 3000–20,000 times [34][35][78,79]. The sequence is bound by the shelterin complex, which is formed by TRF1, TRF2, TPP1, TIN2, POT1 and RAP1 [36][80]. Telomeres are known to maintain the stability of chromosomes and protect genes [37][38][81,82]. Chromosomes without this capping structure, meanwhile, become truncated and fuse with neighboring chromosomes [39][83]. In the process of chromosome replication, the synthesis of Okazaki fragments requires that RNA primers attach to the lagging strand. The shedding RNA then causes telomere shortening. As such, the accumulative loss of telomeres can eventually cause cell cycle arrest and apoptosis, leading to speculation that the progressive reduction in telomere length may play a key role in determining the timing of in vitro cellular senescence [40][41][84,85]. Therefore, telomeres are also viewed by some as a sort of biological clock that controls normal cell proliferation [42][86]. It is estimated that the human telomere loses approximately 24.8 to 27.7 base pairs per year [43][44][87,88]. There are several factors, however, that affect the rate of telomere shortening, including the host’s age [45][89]; gender [46][90]; genetic and epigenetic regulation [47][48][49][91,92,93]; social and economic background [50][51][94,95], and life style factors, such as the lack or presence of exercise, obesity, smoking, and unhealthy diets [44][51][52][88,95,96] (Figure 12). Individuals with shorter telomeres are known to be associated with various age-related diseases and conditions, such as heart failure [53][97], coronary heart disease [54][55][56][98,99,100], diabetes [57][101], osteoporosis [58][102], and a shorter lifespan [59][60][61][103,104,105]. Although the shorter length of telomeres is generally thought to be a marker of poor health and aging, it can lead to genomic instability [62][63][64][106,107,108] and elevated telomerase activity (TA) [65][66][109,110], resulting in a potential cancer predisposition factor.
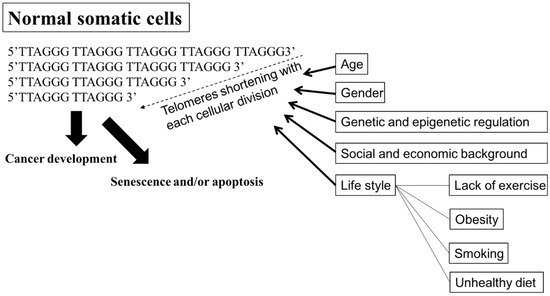
Figure 12. Factors affecting the rate of shortening telomeres. Telomeres are regions of repetitive nucleotide sequences, TTAGGG. Telomeres shorten in each cell division due to incomplete DNA replication. When telomere length progressively reduces to a critical point, the cell then executes senescence or apoptosis, or develops cancer. Several factors, including age; gender; genetic and epigenetic regulation; social and economic background; and life style, such as lack of exercise, obesity, smoking, and an unhealthy diet, are associated with increasing the rate of losing telomeres, leading to senescence or apoptosis, premature death, and cancer development. Bold arrow: shortening DNA leading to senescence and/or apoptosis, or cancer development. Arrow: factors affecting telomeres shortening.
Cell division leads to progressive telomere shortening, resulting in cell senescence; however, the shortening of telomeres can be counteracted by telomerase [67][111]. Telomerase, an RNA-dependent DNA polymerase, is expressed in developing embryos, in reproductive cells (i.e., proliferating germlines), in activated immune cells, and transiently, in adult stem cells, but telomerase is turned off in most adult human tissues. In immortalized cells, telomere length remains stable, with the activation of telomerase being considered one of the main mechanisms underlying this stability [67][68][69][111,112,113]. TA is exhibited in almost all human tumors and in tumor-derived cell lines, while most human somatic cells do not display TA, except for highly proliferative cells, such as bone marrow cells [70][71][114,115]. Telomerase, which is made up of TERT, TERC, and specialized proteins (e.g., dyskerin), preserves telomere stability by adding TTAGGG repeats to the end of the given chromosome (capping) in rapidly dividing cells [68][69][72][112,113,116], using its complementary TERC sequence as the template [73][117], together with TERT subunit as the catalytic component [67][111]. Activities of TERT are frequently up-regulated in human cancers, which is thought to be a critical mechanism contributing to human tumorigenesis [74][75][118,119]. Studies have identified two cancer-specific TERT promoter mutations (C228T and C250T) [76][44] in the activation of telomerase in various cancer cells [77][78][120,121], including GBM [79][122]. The two mutations in the TERT, which cause TERT activation to increase TA to elongate telomere length [76][80][44,123], lead to the proliferative, anti-senescence, and immortal properties of tumor cells. Mutations in the TERT promoter have been detected in more than 50% of primary adult GBM, and these mutations are correlated with EGFR, IDH1, IDH2, TP53, and ATX mutations and increased TA [81][80][82][83][84][45,123,124,125,126]. Mutations in the TERT promoter have been detected in 3–7% of pediatric GBM [76][80][44,123] and tumor cells in this group maintain or increase telomere length through alternative lengthening of telomeres (ALT) pathway [85][86][127,128].
5. Alternative Lengthening of Telomeres, α-Thalassemia X-Linkedintellectual Disability, and Death-Domain-Associated Protein
TA was detected in 76% of cervical cancer cases [87][129], 54% of medullary thyroid cancer cases [88][130], 42% of well-differentiated papillary thyroid cancer cases [89][131], 86.6% of non-small cell lung cancer cases [90][132], and > 80% of hepatocellular carcinoma cases [91][133]. Although telomerase reactivation is the most common mechanism of telomeric repeat addition in cancers [65][109], there is a convincing argument that TA is not the only way for tumors to become immortalized; otherwise, these tumors would have shrunk and died unless they managed to boost their TA to maintain their telomere length. Additionally, Indeed, 5–10% cancer cases exploit a telomerase-independent mechanism to elongate their telomeres, a phenomenon that is also known as ALT [92][134]. In addition to immortalized cells and cancer cells, ALT exists in non-neoplastic tissues, in endothelial, stromal, and some epithelial cells [93][135]. Although the prevalence of the ALT phenotype in cancers is low, ALT is common in certain cancer subtypes, including gliomas [94][136]. While primary GBM has been found to employ telomerase activation, nearly 75% of WHO grades II–III astrocytomas and secondary GBMs, with normal telomerase expression and WT TERT promoter, was observed to employ ALT for the maintenance of telomere length and genome stability [15][95][60,137]. The transformation from telomerase-dependent to ALT-mediated telomere lengthening is considered one of the strategies cancer cells adopt to escape cell senescence and apoptosis caused by telomerase dysfunction or absence [96][97][138,139]. ALT, which is not present in normal cells, often begins with a loss of chromatin remodeling proteins in the telomeres, with a resulting DNA damage response, recombination, and abnormal protein behavior that initiates ALT [98][140].
α-thalassemia X-linked intellectual disability (ATRX) syndrome is characterized by distinctive craniofacial features, genital anomalies, severe developmental delays, hypotonia, intellectual disability, and mild-to-moderate anemia secondary to α-thalassemia [99][141].The ATRX gene, which encodes a SWItch (SWI)/sucrose non-fermenting(SNF)-like chromatin remodeling protein, is frequently mutated in a variety of tumors, including adult lower-grade gliomas, pediatric GBM pediatric adrenocortical carcinoma, osteosarcoma, and neuroblastoma [100][142]. Cancer cells with a loss of ATRX gene display large and bright telomeric DNA foci that are significantly correlated with ALT [85][127], suggesting that ATRX may be a suppressor of the ALT mechanism and a good prognostic factor in cancers, such as in GBMs [101][143]. Moreover, the forced expression of ATRX in ALT-positive and ATRX-negative cell lines abolishes ALT-associated phenotypes [102][103][144,145], but ATRX, either by knocking out or knocking down in telomerase-positive cell lines did not present similar findings [96][103][104][105][106][138,145,146,147,148], suggesting that an ATRX loss alone is not sufficient for ALT activation. DAXX was originally identified as a Fas death receptor binding protein that induced apoptosis via JNK pathway activation. Thus, it was coined the death-domain-associated protein, DAXX [107][149]. ATRX and DAXX were initially seemed to be irrelevant. Analyzing H3.3 chaperone complexes identified ATRX and DAXX [108][109][110][150,151,152]. ATRX, in collaboration with DAXX, deposits H3.3 into telomeric and pericentromeric chromatin to prevent the formation of G-quadruplex DNA (G-4 DNA), a type of DNA structure that promotes homologous recombination repair and DNA-repair mechanisms, leading to telomere shortening [85][102][111][127,144,153]. These results reinforce the value of the ATRX/DAXX/H3.3 complex in ALT suppression (Figure 23). Gliomas with a WT TERT promoter frequently harbor mutations of ATRX to activate ALT [76][44]. Yang et al. successfully switched the telomere lengthening machinery of telomerase-positive cancer cells (HTC75) to an ALT-mediated telomere elongation mechanism by knocking out the TERT, inducing telomeric DNA damage, and disrupting the ATRX/DAXX complex [112][154], suggesting that ATRX and TERT mutations are mutually exclusive in conferring a selective growth advantage in cancers through telomere maintenance. Therefore, effective treatments should not only target TA in cancer cells but should also be aimed atmodulatingthe proper function of the ATRX/DAXX/H3.3 complex to destroy tumor cells.
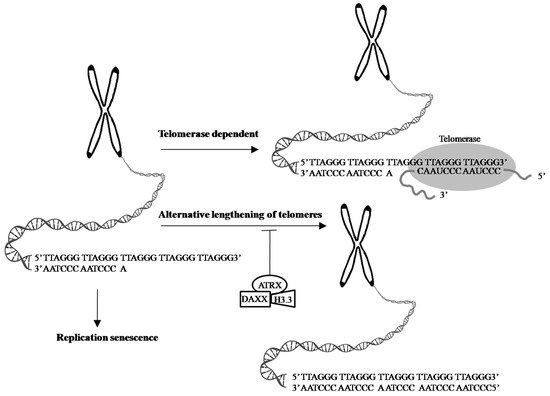
Figure 23. Telomeres are cap-like features at the ends of chromosomes that help protect them when cells divide. Telomeres contain thousands of repeats of the six-nucleotide sequence TTAGGG at the chromosome ends, with complementary DNA strand sequences AATCCC. 1. In the process of cell division, chromosome replication causes progressive telomere shortening, resulting in cell cycle arrest and apoptosis, leading to cellular senescence. 2. The shortening of telomeres can be counteracted by telomerase, which is made up of TERT, TERC, and specialized proteins. Telomerase maintains the length of telomeres stability by adding TTAGGG repeats to the end of the given chromosome, using its complementary TERC sequence as the template, together with TERT subunit as the catalytic component. 3. ALT is a phenomenon that 5–10% cancer cases exploit a telomerase-independent mechanism to elongate their telomeres [92][134]. The transformation from telomerase-dependent to ALT-mediated telomere lengthening may be one of strategies cancer cells adopt to escape cell senescence and apoptosis caused by telomerase dysfunction or absence [96][97][138,139]. ATRX, in collaboration with DAXX and H3.3 promotes the processes of telomere shortening [85][102][111][127,144,153]. Evidence shows the ATRX/DAXX/H3.3 complex in ALT suppression and a good prognostic factor in GBMs [101][143]. Arrow: possible pathway. T-bar: inhibition.