Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Beatrix Zheng and Version 3 by Rok Herman.
by Rok Herman, Nika Aleksandra Kravos, Mojca Jensterle, Andrej Janež and Vita Dolžan
Metetformin is the most commonly used treatment to increase insulin sensitivity in insulin-resistant (IR) conditions such as diabetes, prediabetes, polycystic ovary syndrome, and obesity. This stopic reviewudy explores the underlying mechanisms of the metformin's effect on IR mainly through the changes in the glucose transporter 4 (GLUT4)-mediated glucose transport.
- metformin
- insulin resistance
- glucose transport
- glucose transporter 4
1. Introduction
Several chronic conditions accompanied by insulin resistance (IR) as their main unifying characteristic have been on a steady incline over the last decades [1,2][1][2]. Our understanding of insulin-stimulated glucose transport has advanced enormously over time, from the first report in 1939 that insulin stimulates glucose uptake into rodent muscle, to the comprehension of the importance of glucose transporter translocation after insulin stimulation, to later findings that many different transporters regulate glucose transport into cells [1,3][1][3]. In 1988, James et al. provided the first evidence of the antigenically unique insulin-regulatable glucose transporter protein, which is now widely known as glucose transporter 4 (GLUT4) and is encoded in humans by the solute carrier SLC2A4 gene [4]. In addition to providing considerable insight into vesicle transport and the insulin-signaling pathway, the discovery of GLUT4 has advanced our comprehension of tissue and organ metabolism [1]. With the later findings that GLUT4-mediated transmembrane glucose transport is the rate-limiting step in peripheral glucose utilization and that the level of glucose transporter directly influences glucose disposal in target tissues, the understanding of insulin-stimulated glucose uptake and the role of its deterioration in the states of IR advanced rapidly [5,6,7,8,9][5][6][7][8][9].
In fact, many animal and human studies have indicated that impaired GLUT4-mediated glucose uptake is the primary underlying mechanism of IR. For example, in obese hypertensive rats, representing an animal model of metabolic syndrome, Leguisamo et al. found a reduction in GLUT4 expression accompanied by whole-body IR [10]. Similarly, about 50% lower GLUT4 levels were noted in skeletal muscle of hypertriglyceridemic IR rats compared to normal Wistar rats [11]. In a study on diabetic mouse adipocytes, GLUT4 levels were reduced by 34%, and a 46% reduction in insulin-stimulated glucose transport was observed [12].
Studies in transgenic mouse models facilitated an even better understanding of the role of GLUT4 expression in IR. A 70% decrease in GLUT4 protein expression and a 72% reduction in insulin-stimulated glucose transport resulted from the adipose-specific genetic knockout of GLUT4 [13]. Moreover, the model in this study exhibited IR and glucose intolerance, with significant additional defects in insulin-stimulated glucose uptake in skeletal muscle and insulin-mediated suppression of glucose production in the liver [13]. The same systemic effect of tissue-specific GLUT4 disruption was noted in another experiment by the same research group, in which muscle-specific knockout of GLUT4 expression led to IR in adipose tissue and liver [5]. The research group that conducted both experiments concluded that regardless of which tissue harbors the primary genetic defect of the GLUT4 expression, the other insulin target tissues are ultimately affected [14]. Additional studies on GLUT4 heterozygous knockout mice also showed the development of IR in adipose and muscle tissue and significant enhancement in the likelihood of diabetes development [15,16,17][15][16][17].
On the other hand, the opposite effect was demonstrated in studies on transgenic mice overexpressing GLUT4. In models overexpressing GLUT4 in adipose tissue, a 2-fold in vivo increase in insulin-stimulated glucose transport was noted [18]. Furthermore, Carvalho et al. discovered that overexpression of GLUT4 in adipose tissue of mice with muscle GLUT4 knockout normalized fasting hyperglycemia and glucose intolerance without restoring glucose transport in muscle [19]. Two additional studies came to the same conclusions, but overexpression of GLUT4 improved glucose tolerance and insulin sensitivity in normal as well as genetically diabetic mice [20,21][20][21]. Moreover, transgenic mice engineered to express the human GLUT4 gene and promoter remained highly insulin sensitive on a high-fat diet compared to their nontransgenic counterparts [22].
Human studies have complemented the results obtained in animal models and further emphasized the role of GLUT4 in IR [1]. In 1991, Garvey et al. observed significantly lower GLUT4 mRNA levels in adipose tissue of obese subjects compared to lean controls, and they were even further reduced in the non-insulin-dependent type 2 diabetes mellitus (T2DM) group [23]. Two years later, Rosenbaum et al. published a paper in which they reported that the maximal insulin-stimulated increment in adipocyte glucose transport was independently decreased by obesity and polycystic ovary syndrome (PCOS) compared to controls [24]. In both conditions, GLUT4 content in adipocyte membranes was reduced, and there was a high correlation between the GLUT4 content and the maximum rate of insulin-stimulated glucose uptake in adipocytes [24]. Garvey et al. also studied nondiabetic and diabetic subgroups and demonstrated that human IR involves a defect in GLUT4 trafficking and targeting, leading to the inability of insulin to recruit GLUT4 to the plasma membrane [25]. Furthermore, another study demonstrated the relationship between the severity of IR and GLUT4 expression in skeletal muscle of T2DM patients [26].
Lifelong physical activity was also shown to prevent aging-associated IR in human skeletal muscle myotubes by increasing GLUT4 mRNA and protein expression as well as its translocation ability [27]. Another example of the role of GLUT4 in IR was a study in young, healthy male subjects who completed seven days of bed rest and showed significant IR in muscle biopsies that mainly occurred through reduced GLUT4 content [28]. More recently, Ezeh et al. noted that patients with PCOS had higher scores on the homeostatic model assessment for IR (HOMA-IR) and a trend toward lower GLUT4 mRNA expression in adipose tissue than the healthy control participants [29]. In another paper, the same research group also compared GLUT1 and GLUT4 mRNA levels in adipocytes of PCOS women and matched controls. Their results suggest that IR secondary to lower insulin-mediated glucose uptake and enhanced insulin secretion in PCOS is partly attributable to a reduction in adipocyte GLUT4 expression that is not accompanied by a compensatory increase in GLUT1 expression [30].
2. The Effect of Metformin Treatment on Insulin Resistance
Metformin is a widely prescribed insulin-sensitizing agent in current clinical use. Despite being most commonly associated with its ability to decrease plasma glucose levels, the knowledge about its novel properties and effects continues to evolve [31]. Its action modulates multiple biological pathways. Regarding glucose homeostasis, it mainly works by improving insulin-mediated suppression of hepatic glucose production and enhancing insulin-stimulated glucose disposal in peripheral tissues; however, the precise molecular mechanisms of this action remain elusive [32]. By deploying different measurements for insulin sensitivity in humans, metformin has been shown to improve whole-body sensitivity to insulin in numerous studies in different IR conditions and study groups [33,34,35,36,37,38,39,40,41,42,43][33][34][35][36][37][38][39][40][41][42][43].
Experimental studies have shown that metformin-mediated improvements in insulin sensitivity might be associated with several mechanisms, including increased insulin receptor tyrosine kinase activity, enhanced glycogen synthesis, and at the most downstream end, an increase in the recruitment and activity of GLUT4 [32]. These effects may be achieved through direct and indirect mechanisms [32,44][32][44]. Its effect on several steps in the insulin signaling cascade involved in the generation and propagation of the insulin signal could be one mechanism of overcoming molecular defects of insulin action in IR [32]. Additionally, through inhibition of mitochondrial complex I and other potential pathways, metformin may activate adenosine 5′-monophosphate-activated protein kinase (AMPK), a so-called cellular energy sensor [44,45][44][45]. It also has the ability to stimulate glucagon-like-peptide-1 release, thereby enhancing insulin secretion and lowering plasma glucose levels [46]. Moreover, recent studies suggested that gut microbiota may be one additional target site [47].
Many studies have noted that metformin affects GLUT4 expression, translocation, and function but have not explained the underlying molecular mechanisms. In 1993, Kozka et al. observed that the downregulation of GLUT4 in cultured rat adipocytes after chronic insulin treatment was alleviated by metformin [48]. In a similar study on rat adipocytes, IR induced by chronic insulin treatment was reversed by supplementing metformin in the culture medium [49]. Additionally, in metabolic syndrome-induced rats, 4-week treatment with metformin resulted in a significant decrease in IR and a significant increase in GLUT4 expression in a mixture of heart, liver, and visceral adipose tissues [50]. In a similar study protocol in diabetic mice, metformin treatment improved insulin sensitivity in vitro and increased the expression of GLUT4 in skeletal muscle [51].
Many human studies have come to similar conclusions. In support, Grisouard et al. demonstrated that in vitro 24 h incubation with metformin significantly increased glucose uptake, GLUT4 mRNA expression, and GLUT4 content in the adipocyte plasma membrane [52]. Another in vitro study observed that chronic exposure of cultured human myotubes to metformin was associated with direct enhancement of insulin action, and the increase was correlated with increased GLUT4 mRNA expression [53]. Furthermore, in a study including 35 women with PCOS who received either metformin or rosiglitazone for 6 months, GLUT4 mRNA expression in adipose tissue increased significantly in both groups, with a significant improvement in HOMA-IR [54].
On the other hand, Ciaraldi et al. conducted a study on T2DM subjects who failed glyburide therapy and were randomized to receive additional treatment with metformin or troglitazone for 3–4 months [55]. Metformin treatment increased insulin-stimulated whole-body glucose disposal rates by 20%; however, it did not affect GLUT1 and GLUT4 levels in adipocytes [55]. Another study examined the effect of 26-week metformin treatment in skeletal muscle of moderately obese subjects with newly diagnosed T2DM and found no differences in the insulin-mediated whole-body and leg muscle glucose uptake and unchanged expression of GLUT4 mRNA [56].
Interesting results were obtained in studies investigating GLUT4 expression in the endometria of PCOS patients, which is another example of tissue with IR [57]. For instance, a study by Carvajal et al. measured higher GLUT4 mRNA and protein levels in the endometria of PCOS patients taking metformin for at least 3 months than in patients not on metformin, reaching similar levels to those in the control group [58]. Additional evidence that metformin may improve endometrial IR was provided in a study where obese women with PCOS were given metformin for 3 months and were then compared to obese subjects without PCOS [59]. The endometrial GLUT4 protein and mRNA levels were lower in the PCOS group than in the control group, and the expression of both significantly increased after metformin treatment but was still lower than that of the control group [59]. A few years later, Li et al. observed downregulated levels of endometrial GLUT4 in PCOS conditions, and further in vitro experiments supported the results from previous studies that metformin induces GLUT4 expression [60]. Additional perspective can be gained from a study reporting that metformin could reverse the decreased endometrial GLUT4 expression caused by high testosterone levels (simulating hyperandrogenemia in PCOS) in cultured endometrial cells from non-PCOS women [61].
3. Underlying Mechanisms of the Metformin’s Effect
The underlying mechanisms behind the observed effects of metformin treatment on GLUT4-mediated glucose transport can be divided into changes in insulin signaling pathway mediators, AMPK activation, epigenetic modifications, and enhancements in GLUT4 trafficking and translocation to the plasma membrane. The specific mechanisms are explained in detail in the following sections, and the schematic presentation can be seen in Figure 1.
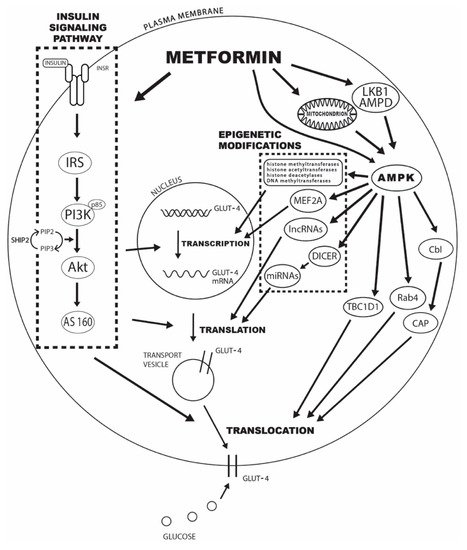
Figure 1. The potential mechanisms of metformin’s effect on increased GLUT4-mediated glucose transport include changes in insulin signaling pathway mediators, AMPK activation, epigenetic modifications, and enhancements in GLUT4 trafficking and translocation to the plasma membrane. Legend: INRS—insulin receptor; IRS—insulin receptor substrate; PI3K—phosphatidylinositol 3-kinase; Akt—protein kinase B; AS160—Akt substrate of 160 kDa; SHIP2—inositol-5-phosphatase 2 with Src homologous domain 2; PIP2—phosphatidylinositol-3,4-diphosphate; PIP3—phosphatidylinositol-3,4,5-triphosphate; AMPD—AMP deaminase; AMPK—adenosine 5′-monophosphate-activated protein kinase; MEF2A—myocyte enhancer 2A; lncRNAs—long noncoding RNAs; miRNAs—microRNAs; TBC1D1—TBC domain family, member 1; CAP—Cbl-associated protein.
3.1. Insulin Signaling Pathway
The insulin signaling pathway needs to be explored when analyzing the effects of metformin treatment on glucose uptake. The binding of the insulin molecule to the α subunit of the insulin receptor (INSR) causes its dimerization and the activation of the intrinsic kinase activity of the β subunits, leading to their autophosphorylation. Activated INSR acts on adaptor proteins from the insulin receptor substrate family (IRS1, IRS2, and IRS3) in a further cascade. Of particular importance are IRS1 and IRS2, which bind to the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K) and activate it through phosphorylation. Lipid products of PI3K catalyze the formation of phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 then binds to phosphatidylinositol-3,4,5-phosphate kinase 1 (PDK1) and protein kinase B (Akt) and attracts Akt to the cell membrane, where PDK1 phosphorylates Akt at Thr308. Additional phosphorylation at Ser473/474 is required for the complete activation of Akt. Activated Akt, through its mediator Akt substrate of 160 kDa (AS160), represents a crucial step in the regulation of the kinase cascade involved in the translocation of GLUT4 from intracellular transport vesicles to the plasma membrane [23,31,32,62][23][31][32][62]. By studying the effects of their inhibition and overexpression, a lot of evidence has been obtained suggesting that PI3K and Akt activation are essential for insulin-stimulated GLUT4 translocation [63,64][63][64]. The initial stages of the signaling pathway, especially signaling via IRS, PI3K, and Akt, have been described as crucial for the development of IR, and decreased expression of PI3K, Akt, and GLUT4 has been reported in different tissues of IR patients [32,55,65,66,67,68,69][32][55][65][66][67][68][69].
One of the first studies of metformin’s effect on insulin signaling mediators demonstrated that chronic insulin treatment reduced INRS tyrosine phosphorylation, PI3K activity, and Akt activity by 60–70% in cultured rat adipocytes, and these effects were prevented by the inclusion of metformin in the culture medium [49]. A later study in C2C12 cells reached similar conclusions and suggested that chronic insulin treatment drastically reduced insulin-stimulated tyrosine phosphorylation of INRS and IRS1 as well as PI3K activity, and treatment with metformin was able to reverse these changes [70]. Rice et al. demonstrated that incubation with metformin increased IRS1 mRNA and protein expression in human granulosa cell culture, as well as IRS2 protein expression [71]. In this study, as well as in a similar study by Sonntag et al., metformin incubation contributed to the additional activation of Akt in the presence of insulin via PI3K activation [71,72][71][72]. Furthermore, in ovarian tissue of rats exposed to a hyperandrogenic environment in fetal development with consequent ovulation disorders, markedly lower levels of INSR, IRS1, IRS2, and GLUT4 mRNA and proteins were found, and incubation with metformin increased mRNA and protein expression and the activation of most of these mediators [73]. Another study using the rat PCOS model demonstrated a significant reduction in IRS2 and PI3K expression, and 4-week in vivo metformin treatment significantly increased their expression [74]. Furthermore, in an additional study, reduced PI3K and Akt expression observed in ovarian and hepatic tissues of the rat PCOS model increased significantly after metformin treatment [75]. In a study by Ferreira et al., both metformin and insulin increased PI3K and GLUT4 expression in endometrial cell culture, and their combined effect was even more pronounced [67]. In contrast to insulin, metformin reduced the rate of Akt phosphorylation at Ser473 in this study [67].
However, some studies reported opposing results. For example, in a study by Ma et al., 24 h incubation of human granulosa cell culture with metformin resulted in reduced IRS1 mRNA and protein expression [76]. Moreover, in a study by Ciaraldi et al., unchanged levels of IRS1, GLUT1, GLUT4, p85, and Akt proteins were observed in adipocytes of patients with T2DM after 3-4 months of metformin treatment [55]. Similarly, insulin signaling parameters were unchanged after 26 weeks of metformin treatment in newly diagnosed T2DM patients [56]. Furthermore, Kim et al. studied the effect of 3-4 months of metformin treatment in T2DM subjects who failed glyburide treatment and noted no effect on basal or insulin-stimulated IRS1-associated PI3K or Akt activity in muscle cells. Additionally, protein expression of IRS1, the p85 subunit of PI3K, and Akt was unaltered after treatment [77].
Additional perspective can be gained from an in vitro study of the effects of metformin on human podocytes and a mouse skeletal muscle cell line, which demonstrated an essential role for inositol-5-phosphatase 2 with Src homologous domain 2 (SHIP2) [78]. SHIP2 hydrolyzes PIP3 back to phosphatidylinositol-3,4-diphosphate (PIP2), which is the opposite of the action of PI3K in the insulin signaling pathway. SHIP2 is overexpressed in IR tissues, and in this study, direct binding of metformin to SHIP2 and decreased activity of the phosphatase domain of SHIP2 were found [78]. At the same time, no effect of metformin was observed on the level of Akt phosphorylation [78].
3.2. AMPK Activation
AMPK is a serine/threonine-specific protein kinase, acting as a highly conserved master regulator of metabolism [79]. It is regulated by multiple upstream signals and affects numerous downstream substrates [80]. It exists as a trimeric complex consisting of a catalytic subunit (α-subunit) and two regulatory subunits (β- and γ-subunits) [80]. The N-terminus of the α-subunit comprises the kinase domain, and the phosphorylation of a conserved threonine (referred to as Thr172) in the kinase domain is required for the full activation of AMPK [80]. An increase in AMPK activity was associated with quickly increased glucose uptake and translocation of GLUT4 to the plasma membrane [52,63,81,82,83,84,85,86,87,88][52][63][81][82][83][84][85][86][87][88]. Zhou et al. were one of the first groups to propose AMPK phosphorylation and activation as a critical pathway for metformin’s pleiotropic effects [63]. The hypothesis of the metformin–AMPK–GLUT4 pathway was established; however, the exact mediators remain the subject of debate. Although metformin treatment has been coupled to AMPK activation and upregulation of glucose uptake separately, only a few studies have measured both AMPK activation and glucose uptake [63,89,90,91,92,93][63][89][90][91][92][93].
One of the most frequently invoked mechanisms of metformin action is the inhibition of mitochondrial complex I and the consequent reduction in the [ATP]:[ADP] and [ATP]:[AMP] ratios [80,82][80][82]. Due to the transient changes in cellular energy status, ATP is replaced with either ADP or AMP, allosterically activating AMPK [82,94][82][94]. Metformin has also been shown to activate AMPK through phosphorylation at Thr172 of the α-subunit independently of changes in adenine nucleotides [82,95,96,97][82][95][96][97]. In addition, LKB1 was identified as an upstream kinase responsible for phosphorylating and activating AMPK and was implicated as a major target of metformin [82,98,99][82][98][99]. However, metformin probably does not directly activate either LKB1 or AMPK since it did not influence the phosphorylation of AMPK by LKB1 in a cell-free assay [63,100][63][100]. Metformin may also interact directly with the γ-subunit of the AMPK complex, producing a structural change that promotes activation [101]. In a more novel approach, inhibition of AMP deaminase (AMPD) has been proposed as alternative mechanism to AMPK activation [102], and one report suggested that metformin may control AMP levels via effects on AMPD [103].
A study on mouse soleus muscle showed that chronic, but not acute, in vivo metformin treatment enhanced insulin-stimulated glucose uptake through activation of AMPK without notable changes in protein expression of insulin signaling mediators or GLUT4 [89]. An in vitro study in human adipocytes obtained from surgical biopsies showed that metformin incubation for 24 h increased glucose uptake, GLUT4 mRNA expression, and cellular protein level; however, suppression of metformin-induced AMPK activity by AMPKα1 silencing reduced the observed effects [52]. An approximately twofold increase in AMPK activity, independent of the AMPK level or glycemia, was also shown in adipose tissue of patients with T2DM treated with metformin for 10 weeks compared with gliclazide therapy [104]. The amount of GLUT4 protein was unaltered, and metformin also did not significantly alter the levels of insulin signaling pathway mediators [104]. These results were additionally confirmed in 3T3-L1 adipocytes [104]. However, at the time of recruitment, 12 out of 20 participants were already taking metformin, and the study had a six-week run-in period when medication was discontinued. The duration of this run-in period is important in light of a recent study that concluded that six months after metformin withdrawal, GLUT4 mRNA expression in subcutaneous adipose tissue of PCOS patients remained stable [105]. Another study in patients with T2DM showed that metformin treatment for 10 weeks significantly increased AMPK activity in skeletal muscle, and the observed effect was associated with phosphorylation of AMPK at Thr172 and resulted in enhanced peripheral glucose uptake [92]. Furthermore, in the endometria of PCOS patients, metformin increased GLUT4 mRNA and protein levels [58]. It significantly increased the phosphorylation and, therefore, the activity of both AMPKα and myocyte enhancer 2A (MEF2A), a transcription factor with a known binding site on the promoter region of the GLUT4 gene [58,106][58][106].
Some studies have also demonstrated the importance of metformin’s action through AMPK activation and its downstream effect on GLUT4 translocation. Activated AMPK can phosphorylate TBC1D1 (TBC domain family, member 1), which controls the translocation and plasma membrane levels of GLUT4 [80,107][80][107]. Lee et al. showed the critical role of AMPK in GLUT4 translocation in the C2C12 skeletal muscle cell line [108]. Two years later, Lee and colleagues published another study demonstrating the importance of AMPK in GLUT4 translocation in 3T3-L1 preadipocyte cells, and this effect was attenuated by AMPK knockdown [109].
3.3. Epigenetic Modifications
In an effort to better understand the etiology of IR conditions, the focus has now shifted from genome-wide and candidate gene association studies to the epigenetic mechanisms that could explain the dynamic and well-observed interplay between known genetic and environmental factors [110,111,112][110][111][112]. Therefore, a hypothesis that at least some of metformin’s effects are mediated through different epigenetic modifications was proposed, and the evidence supporting it has grown in recent years.
Treatment with metformin might influence the activity of numerous epigenetic modifying enzymes, mainly through the promotion of phosphorylation and hence activation of AMPK [113,114][113][114]. Activated AMPK can phosphorylate multiple epigenetic enzymes, such as histone acetyltransferases, class II histone deacetylases, and DNA methyltransferases [113]. For example, a study by McGee et al. reported that AMPK activation decreased the transcriptional repressor histone deacetylase 5 (HDAC5), with a known association with the GLUT4 gene, resulting in increased GLUT4 expression in human myotubes [115]. In addition, metformin has also been reported to decrease the expression of multiple histone methyltransferases and to increase the activity of the class III histone deacetylase SIRT1 [113]. The importance of such mechanisms can be seen in a study in which 3T3-L1 adipocytes with knocked down SIRT1 showed inhibited insulin-stimulated glucose uptake and impaired GLUT4 translocation [116].
Furthermore, due to the consistent findings of the importance of posttranscriptional modifications of GLUT4 mRNA and the previously established association of altered microRNA (miRNA) levels with diabetes, IR, and inflammation, the effect of metformin on various miRNAs has also been studied in recent years [117,118][117][118]. miRNAs are small noncoding regulatory RNAs that act as negative posttranscriptional regulators of gene expression [119,120][119][120]. Alterations of miRNA expression by metformin may be partially explained by an increase in DICER, one of the critical enzymes in miRNA processing [113]. Increases in DICER protein levels have been reported in metformin-treated diabetic humans and mice and also in cells treated with the direct AMPK activator 5-aminoimidazole-4-carboxamide riboside (AICAR), suggesting that AMPK activation is the primary mechanism [113,121,122,123][113][121][122][123].
Numerous miRNAs have been reported to directly or indirectly regulate insulin sensitivity and GLUT4 expression, and their levels were shown to be altered in IR conditions [101,119,124,125,126,127,128,129][101][119][124][125][126][127][128][129]. Treatment with metformin has been associated with changes in several miRNAs, with many of them having a known effect on glucose metabolism. In a model of high-fat diet rats and IR skeletal muscle L6 cells, metformin dose-dependently decreased miR-21 expression, consequently improving skeletal muscle IR [130]. A recent study examined the effect of metformin on miR-223 expression and the amount of Akt and GLUT4 proteins in IR 3T3-L1 adipocyte cells and adipocytes of diabetic patients [101]. MiR-223 was overexpressed in both, and incubation of the cell line and 3-month treatment of patients with metformin decreased its expression while increasing Akt and GLUT4 expression [101]. Mensà et al. found that miR-146a levels show a significant age-related decline that is even more notable in T2DM patients [131]. They also observed that miR-146a was significantly overexpressed in T2DM patients treated with metformin [131]. Although miR-146a is more commonly associated with inflammation [132], it has also been associated with IR and poor glycemic control in Asian Indian T2DM patients [133]. Furthermore, a randomized, double-blinded, and placebo-controlled three-month trial of metformin treatment in T2DM patients found that metformin (but not placebo) led to significant changes in circulating miR-192, miR-140-5p, and miR-222, in parallel to decreased fasting glucose and HbA1c [134]. More recently, the same concept of metformin’s effect on extracellular miRNAs was demonstrated by Ghai et al. [135]. They observed that the concentrations of several miRNAs were increased in T2DM, but they decreased to the normal range after metformin treatment. However, at present, there is little evidence that these circulating miRNAs are involved in GLUT4 expression or IR [135]. In a study in PCOS patients, 12-month metformin treatment reduced serum levels of miR-122, miR-223, and miR-29a, all of which have previously been reported to influence glucose metabolism [136].
A new area of research of metformin’s mode of action is likely to expand to other epigenetic mechanisms. In 2020, García-Calzón et al. published a paper where they discovered that by measuring blood-based epigenetic markers in the form of DNA methylation of 11 specific loci in drug-naive patients with T2DM, they could discriminate between glycemic responders and nonresponders to metformin, providing some further evidence into the role of DNA methylation [137]. Additionally, there has been limited research covering the role of long noncoding RNAs (lncRNAs) to date. In C2C12 myotubes, metformin reduced medium glucose concentration in the culture medium, increased levels of GLUT4 in the plasma membrane, and repressed lncRNA Dreh expression [138]. Interestingly, knockdown of Dreh had even more profound results, while its overexpression attenuated the glucose-lowering effect of metformin, suggesting that glucoregulatory actions of metformin are mediated in part by the lncRNA Dreh [138]. In gastric cancer cells, metformin treatment significantly inhibited the cellular functions of cancer cells, and lncRNA H19 was a crucial component in that process [139]. Interestingly, the same lncRNA H19 was previously shown to improve IR in skeletal muscle, although the proposed mechanism involves reducing ectopic lipid accumulation, not GLUT4 expression [140].
3.4. GLUT4 Trafficking and Translocation
Many studies have suggested that defects in GLUT4 translocation are closely related to IR [141,142,143,144,145][141][142][143][144][145]. The stimulation of glucose uptake requires the translocation of GLUT4 from tubulovesicular structures, named GLUT4 storage vesicles (GSV), to the cell surface, rapidly increasing the GLUT4 density at the plasma membrane and consequently glucose uptake [119]. It is well documented that insulin enhances GLUT4 translocation through incompletely defined intracellular signaling pathways involved in releasing GSVs from intracellular retention, their trafficking, tethering, and, finally, docking and fusion to the plasma membrane [7,52,119,146,147,148,149,150][7][52][119][146][147][148][149][150]. Furthermore, studies from the 1990s onwards suggested that metformin ameliorates IR independently of GLUT4 protein synthesis, mainly through subcellular redistribution of GLUT4 [49,147,151,152,153,154,155,156][49][147][151][152][153][154][155][156]. However, the mechanisms by which metformin acts on GLUT4 translocation are still largely unknown.
The understanding of glucose uptake involves the interplay between the insulin signaling pathway and GLUT4 membrane trafficking at the cellular level. Not surprisingly, identifying the molecules that link them has been a significant research focus. Since metformin has a known effect on the insulin signaling pathway, this could also explain the noted effects of metformin treatment on the GLUT4 translocation process [148]. The lipid products of PI3K contribute to the activation of Akt and atypical protein kinases C zeta and lambda (PKCζ/λ) [55,119][55][119]. The discovery in 2003 that a RabGAP, AS160, is a highly insulin-responsive Akt target provided one of the first links between insulin signaling and GLUT4 translocation, given that a significant function of Rab GTPases is to regulate vesicle traffic [1,119,148,157][1][119][148][157]. AS160 has emerged as a negative regulator in the insulin transduction relay since overexpressing a phosphorylation-defective mutant reduced insulin-dependent GLUT4 translocation, and, conversely, deletion of AS160 elevated GLUT4 levels in the plasma membrane in the absence of insulin stimulation [1,158][1][158]. Moreover, Rice et al. demonstrated that metformin increased insulin-stimulated translocation of GLUT4 to the plasma membrane in human ovarian granulosa cells via a mechanism involving PI3K activation of Akt [71]. Additionally, Polianskyte-Prause et al. demonstrated a novel mechanism of metformin action through a reduction in SHIP2 activity, since in SHIP2-overexpressing myotubes, metformin ameliorated reduced glucose uptake by slowing down GLUT4 endocytosis [78]. In addition to this, a step-by-step review of the protein related to the GLUT4 translocation machinery in skeletal muscle reported that several miRNAs have been potentially correlated with some target genes and could present additional targets of metformin action [119].
Some authors also demonstrated the crucial role of AMPK activation and its downstream effect on GLUT4 translocation mediators after metformin treatment. For example, Lee et al. noted metformin’s activity through the phosphorylation of Cbl and stimulated expression of Cbl-associated protein (CAP) in an AMPK-dependent manner, and Cbl/CAP-associated multicomplex formation modulated GLUT4 translocation in 3T3-L1 cells [109]. In another paper, Lee et al. demonstrated that metformin induced Rab4 expression via the AMPK pathway and demonstrated that the activities of AS160 and PKCζ are involved in metformin-induced Rab4 regulation in C2C12 cells [108]. Together, these studies suggest that metformin also induces Rab4 expression via AMPK–AS160–PKCζ and modulates insulin-mediated GLUT4 translocation [108]. On the other hand, Kristensen et al. did not note any changes in insulin signaling upon Akt and AS160 protein expression or their phosphorylation or changes in protein expression of Rab4 after two weeks of metformin treatment in mouse soleus muscle [89].
4. Conclusions
IR is a clinical condition shared by many diseases other than prediabetes and T2DM, such as obesity, PCOS, and nonalcoholic fatty liver disease. Therefore, the comprehension of the mechanisms responsible for impaired insulin action is fundamental in the attempt to ameliorate IR and to account for the favorable effects of insulin sensitizers. Although we do not know how to explain all of the mechanisms of metformin action at the molecular level, metformin, active at all sites of impaired insulin action, has been a first-line drug to improve IR for decades. The observed increase in peripheral glucose utilization after metformin treatment most likely derives from the induction of GLUT4 expression and its enhanced translocation to the plasma membrane. Based on the current data, metformin achieves this effect through direct and indirect effects on mediators from the initial stages of the insulin signaling pathway, AMPK activation, GLUT4 trafficking and translocation mediators, and complex AMPK-dependent and -independent epigenetic modifications. A better understanding of the mechanisms behind its clinically relevant increase in insulin sensitivity could help researchers and clinicians studying and treating the dramatic rise in IR conditions worldwide.
References
- Klip, A.; McGraw, T.E.; James, D.E. Thirty Sweet Years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Prentki, M. Insulin Resistance and Insulin Hypersecretion in the Metabolic Syndrome and Type 2 Diabetes: Time for a Conceptual Framework Shift. Diabetes Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Mueckler, M. Glucose Transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [PubMed]
- James, D.E.; Brown, R.; Navarro, J.; Pilch, P.F. Insulin-Regulatable Tissues Express a Unique Insulin-Sensitive Glucose Transport Protein. Nature 1988, 333, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted Disruption of the Glucose Transporter 4 Selectively in Muscle Causes Insulin Resistance and Glucose Intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Cline, G.W.; Petersen, K.F.; Krssak, M.; Shen, J.; Hundal, R.S.; Trajanoski, Z.; Inzucchi, S.; Dresner, A.; Rothman, D.L.; Shulman, G.I. Impaired Glucose Transport as a Cause of Decreased Insulin-Stimulated Muscle Glycogen Synthesis in Type 2 Diabetes. N. Engl. J. Med. 1999, 341, 240–246. [Google Scholar] [CrossRef]
- Armoni, M.; Harel, C.; Karnieli, E. GLUT4 Goes Abnormal: Disregulation of the Insulin-Responsive Glucose Transporter in Abnormal Metabolic States. Curr. Med. Chem.-Immunol. Endocr. Metab. Agents 2005, 5, 207–217. [Google Scholar] [CrossRef]
- Karnieli, E.; Zarnowski, M.J.; Hissin, P.J.; Simpson, I.A.; Salans, L.B.; Cushman, S.W. Insulin-Stimulated Translocation of Glucose Transport Systems in the Isolated Rat Adipose Cell. Time Course, Reversal, Insulin Concentration Dependency, and Relationship to Glucose Transport Activity. J. Biol. Chem. 1981, 256, 4772–4777. [Google Scholar] [CrossRef]
- Karnieli, E.; Barzilai, A.; Rafaeloff, R.; Armoni, M. Distribution of Glucose Transporters in Membrane Fractions Isolated from Human Adipose Cells. Relation to Cell Size. J. Clin. Investig. 1986, 78, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Leguisamo, N.M.; Lehnen, A.M.; Machado, U.F.; Okamoto, M.M.; Markoski, M.M.; Pinto, G.H.; Schaan, B.D. GLUT4 Content Decreases along with Insulin Resistance and High Levels of Inflammatory Markers in Rats with Metabolic Syndrome. Cardiovasc. Diabetol. 2012, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Seböková, E.; Klimes, I.; Moss, R.; Mitková, A.; Wiersma, M.; Bohov, P. Decreased Glucose Transporter Protein (GLUT4) in Skeletal Muscle of Hypertriglyceridaemic Insulin-Resistant Rat. Physiol. Res. 1995, 44, 87–92. [Google Scholar] [PubMed]
- Napoli, R.; Hirshman, M.F.; Horton, E.S. Mechanisms and Time Course of Impaired Skeletal Muscle Glucose Transport Activity in Streptozocin Diabetic Rats. J. Clin. Investig. 1995, 96, 427–437. [Google Scholar] [CrossRef]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-Selective Targeting of the GLUT4 Gene Impairs Insulin Action in Muscle and Liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.E.; Kahn, B.B. Tissue-Specific Alterations of Glucose Transport and Molecular Mechanisms of Intertissue Communication in Obesity and Type 2 Diabetes. Horm. Metab. Res. Horm. Stoffwechselforsch. Horm. Metab. 2007, 39, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, L.; Stenbit, A.E.; Chen, W.; Hu, M.; Barzilai, N.; Katz, E.B.; Charron, M.J. Peripheral but Not Hepatic Insulin Resistance in Mice with One Disrupted Allele of the Glucose Transporter Type 4 (GLUT4) Gene. J. Clin. Investig. 1997, 100, 1831–1839. [Google Scholar] [CrossRef]
- Stenbit, A.E.; Tsao, T.S.; Li, J.; Burcelin, R.; Geenen, D.L.; Factor, S.M.; Houseknecht, K.; Katz, E.B.; Charron, M.J. GLUT4 Heterozygous Knockout Mice Develop Muscle Insulin Resistance and Diabetes. Nat. Med. 1997, 3, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Houseknecht, K.L.; Stenbit, A.E.; Katz, E.B.; Charron, M.J. Reduced Glucose Uptake Precedes Insulin Signaling Defects in Adipocytes from Heterozygous GLUT4 Knockout Mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, P.R.; Gnudi, L.; Tozzo, E.; Yang, H.; Leach, F.; Kahn, B.B. Adipose Cell Hyperplasia and Enhanced Glucose Disposal in Transgenic Mice Overexpressing GLUT4 Selectively in Adipose Tissue. J. Biol. Chem. 1993, 268, 22243–22246. [Google Scholar] [CrossRef]
- Carvalho, E.; Kotani, K.; Peroni, O.D.; Kahn, B.B. Adipose-Specific Overexpression of GLUT4 Reverses Insulin Resistance and Diabetes in Mice Lacking GLUT4 Selectively in Muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E551–E561. [Google Scholar] [CrossRef]
- Brozinick, J.T.J.; McCoid, S.C.; Reynolds, T.H.; Nardone, N.A.; Hargrove, D.M.; Stevenson, R.W.; Cushman, S.W.; Gibbs, E.M. GLUT4 Overexpression in Db/Db Mice Dose-Dependently Ameliorates Diabetes but Is Not a Lifelong Cure. Diabetes 2001, 50, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Burcelin, R.; Katz, E.B.; Huang, L.; Charron, M.J. Enhanced Insulin Action Due to Targeted GLUT4 Overexpression Exclusively in Muscle. Diabetes 1996, 45, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, B.J.; Griesel, B.A.; King, C.D.; Josey, M.A.; Olson, A.L. Moderate GLUT4 Overexpression Improves Insulin Sensitivity and Fasting Triglyceridemia in High-Fat Diet-Fed Transgenic Mice. Diabetes 2013, 62, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Garvey, W.T.; Maianu, L.; Huecksteadt, T.P.; Birnbaum, M.J.; Molina, J.M.; Ciaraldi, T.P. Pretranslational Suppression of a Glucose Transporter Protein Causes Insulin Resistance in Adipocytes from Patients with Non-Insulin-Dependent Diabetes Mellitus and Obesity. J. Clin. Investig. 1991, 87, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.; Haber, R.S.; Dunaif, A. Insulin Resistance in Polycystic Ovary Syndrome: Decreased Expression of GLUT-4 Glucose Transporters in Adipocytes. Am. J. Physiol.-Endocrinol. Metab. 1993, 264, E161–E166. [Google Scholar] [CrossRef] [PubMed]
- Garvey, W.T.; Maianu, L.; Zhu, J.H.; Brechtel-Hook, G.; Wallace, P.; Baron, A.D. Evidence for Defects in the Trafficking and Translocation of GLUT4 Glucose Transporters in Skeletal Muscle as a Cause of Human Insulin Resistance. J. Clin. Investig. 1998, 101, 2377–2386. [Google Scholar] [CrossRef]
- Kampmann, U.; Christensen, B.; Nielsen, T.S.; Pedersen, S.B.; Ørskov, L.; Lund, S.; Møller, N.; Jessen, N. GLUT4 and UBC9 Protein Expression Is Reduced in Muscle from Type 2 Diabetic Patients with Severe Insulin Resistance. PLoS ONE 2011, 6, e27854. [Google Scholar] [CrossRef] [PubMed]
- Bunprajun, T.; Henriksen, T.I.; Scheele, C.; Pedersen, B.K.; Green, C.J. Lifelong Physical Activity Prevents Aging-Associated Insulin Resistance in Human Skeletal Muscle Myotubes via Increased Glucose Transporter Expression. PLoS ONE 2013, 8, e66628. [Google Scholar] [CrossRef]
- Biensø, R.S.; Ringholm, S.; Kiilerich, K.; Aachmann-Andersen, N.-J.; Krogh-Madsen, R.; Guerra, B.; Plomgaard, P.; van Hall, G.; Treebak, J.T.; Saltin, B.; et al. GLUT4 and Glycogen Synthase Are Key Players in Bed Rest-Induced Insulin Resistance. Diabetes 2012, 61, 1090–1099. [Google Scholar] [CrossRef]
- Ezeh, U.; Chen, I.Y.-D.; Chen, Y.-H.; Azziz, R. Adipocyte Insulin Resistance in PCOS: Relationship With GLUT-4 Expression and Whole-Body Glucose Disposal and β-Cell Function. J. Clin. Endocrinol. Metab. 2020, 105, e2408–e2420. [Google Scholar] [CrossRef]
- Ezeh, U.; Chen, I.Y.-D.; Chen, Y.-H.; Azziz, R. Adipocyte Expression of Glucose Transporter 1 and 4 in PCOS: Relationship to Insulin-Mediated and Non-Insulin-Mediated Whole-Body Glucose Uptake. Clin. Endocrinol. (Oxf.) 2019, 90, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kover, K.L.; Moore, W.V. New Insight into Metformin Mechanism of Action and Clinical Application. In Metformin; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Giannarelli, R.; Aragona, M.; Coppelli, A.; Del Prato, S. Reducing Insulin Resistance with Metformin: The Evidence Today. Diabetes Metab. 2003, 29, 6S28–6S35. [Google Scholar] [CrossRef]
- Pala, L.; Barbaro, V.; Dicembrini, I.; Rotella, C.M. The Therapy of Insulin Resistance in Other Diseases besides Type 2 Diabetes. Eat. Weight Disord. 2014, 19, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kamenova, P. Therapeutic Potential of Metformin in Normal Glucose Tolerant Persons with Metabolic Syndrome. Biotechnol. Biotechnol. Equip. 2020, 34, 30–37. [Google Scholar] [CrossRef]
- Malin, S.K.; Gerber, R.; Chipkin, S.R.; Braun, B. Independent and Combined Effects of Exercise Training and Metformin on Insulin Sensitivity in Individuals with Prediabetes. Diabetes Care 2012, 35, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Worsley, R.; Jane, F.; Robinson, P.J.; Bell, R.J.; Davis, S.R. Metformin for Overweight Women at Midlife: A Double-Blind, Randomized, Controlled Trial. Climacteric 2015, 18, 270–277. [Google Scholar] [CrossRef]
- Nawrocka-Rutkowska, J.; Ciecwiez, S.; Marciniak, A.; Brodowska, A.; Wiśniewska, B.; Kotlega, D.; Starczewski, A. Insulin Resistance Assessment in Patients with Polycystic Ovary Syndrome Using Different Diagnostic Criteria–Impact of Metformin Treatment. Ann. Agric. Environ. Med. 2013, 20, 528–532. [Google Scholar]
- Bjornstad, P.; Schäfer, M.; Truong, U.; Cree-Green, M.; Pyle, L.; Baumgartner, A.; Garcia Reyes, Y.; Maniatis, A.; Nayak, S.; Wadwa, R.P.; et al. Metformin Improves Insulin Sensitivity and Vascular Health in Youth With Type 1 Diabetes Mellitus. Circulation 2018, 138, 2895–2907. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, M.; Payami, S.; Payami, S.; Ashtary-Larky, D.; Nikzamir, A.; Mohammadzadeh, G. Evaluation of Insulin Resistance Indices in Type 2 Diabetic Patients Treated with Different Anti-Diabetic Drugs. Open J. Endocr. Metab. Dis. 2016, 6, 95–101. [Google Scholar] [CrossRef]
- Agarwal, S.M.; Panda, R.; Costa-Dookhan, K.A.; MacKenzie, N.E.; Treen, Q.C.; Caravaggio, F.; Hashim, E.; Leung, G.; Kirpalani, A.; Matheson, K.; et al. Metformin for Early Comorbid Glucose Dysregulation and Schizophrenia Spectrum Disorders: A Pilot Double-Blind Randomized Clinical Trial. Transl. Psychiatry 2021, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Atabek, M.E.; Pirgon, O. Use of Metformin in Obese Adolescents with Hyperinsulinemia: A 6-Month, Randomized, Double-Blind, Placebo-Controlled Clinical Trial. J. Pediatr. Endocrinol. Metab. 2008, 21, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Fux Otta, C.; Wior, M.; Iraci, G.S.; Kaplan, R.; Torres, D.; Gaido, M.I.; Wyse, E.P. Clinical, Metabolic, and Endocrine Parameters in Response to Metformin and Lifestyle Intervention in Women with Polycystic Ovary Syndrome: A Randomized, Double-Blind, and Placebo Control Trial. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2010, 26, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Brufani, C.; Crinò, A.; Fintini, D.; Patera, P.I.; Cappa, M.; Manco, M. Systematic Review of Metformin Use in Obese Nondiabetic Children and Adolescents. Horm. Res. Paediatr. 2013, 80, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, M.P.; Marek, B.; Kajdaniuk, D.; Rokicka, D.; Szymborska-Kajanek, A.; Strojek, K. Metformin-A New Old Drug. Endokrynol. Pol. 2017, 68, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. (Lausanne) 2020, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the Gastrointestinal Tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hu, N. Effects of Metformin on the Gut Microbiota in Obesity and Type 2 Diabetes Mellitus. Diabetes. Metab. Syndr. Obes. 2020, 13, 5003–5014. [Google Scholar] [CrossRef] [PubMed]
- Kozka, I.J.; Holman, G.D. Metformin Blocks Downregulation of Cell Surface GLUT4 Caused by Chronic Insulin Treatment of Rat Adipocytes. Diabetes 1993, 42, 1159–1165. [Google Scholar] [CrossRef]
- Pryor, P.R.; Liu, S.C.; Clark, A.E.; Yang, J.; Holman, G.D.; Tosh, D. Chronic Insulin Effects on Insulin Signalling and GLUT4 Endocytosis Are Reversed by Metformin. Biochem. J. 2000, 348, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Zayed, E.A.; AinShoka, A.A.; El Shazly, K.A.; Abd El Latif, H.A. Improvement of Insulin Resistance via Increase of GLUT4 and PPARγ in Metabolic Syndrome-Induced Rats Treated with Omega-3 Fatty Acid or l-Carnitine. J. Biochem. Mol. Toxicol. 2018, 32, e22218. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, L.; Zhou, N.; Gai, S.; Liu, X.; Zhang, S. Beneficial Effects of Combination Therapy of Phloretin and Metformin in Streptozotocin-Induced Diabetic Rats and Improved Insulin Sensitivity in Vitro. Food Funct. 2020, 11, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Grisouard, J.; Timper, K.; Radimerski, T.M.; Frey, D.M.; Peterli, R.; Kola, B.; Korbonits, M.; Herrmann, P.; Krähenbühl, S.; Zulewski, H.; et al. Mechanisms of Metformin Action on Glucose Transport and Metabolism in Human Adipocytes. Biochem. Pharmacol. 2010, 80, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalili, L.; Forsgren, M.; Kannisto, K.; Zierath, J.R.; Lönnqvist, F.; Krook, A. Enhanced Insulin-Stimulated Glycogen Synthesis in Response to Insulin, Metformin or Rosiglitazone Is Associated with Increased MRNA Expression of GLUT4 and Peroxisomal Proliferator Activator Receptor Gamma Co-Activator 1. Diabetologia 2005, 48, 1173–1179. [Google Scholar] [CrossRef]
- Jensterle, M.; Janez, A.; Mlinar, B.; Marc, J.; Prezelj, J.; Pfeifer, M. Impact of Metformin and Rosiglitazone Treatment on Glucose Transporter 4 MRNA Expression in Women with Polycystic Ovary Syndrome. Eur. J. Endocrinol. 2008, 158, 793–801. [Google Scholar] [CrossRef]
- Ciaraldi, T.P.; Kong, A.P.S.; Chu, N.V.; Kim, D.D.; Baxi, S.; Loviscach, M.; Plodkowski, R.; Reitz, R.; Caulfield, M.; Mudaliar, S.; et al. Regulation of Glucose Transport and Insulin Signaling by Troglitazone or Metformin in Adipose Tissue of Type 2 Diabetic Subjects. Diabetes 2002, 51, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.K.R.; Hällsten, K.; Björnholm, M.; Tsuchida, H.; Chibalin, A.V.; Virtanen, K.A.; Heinonen, O.J.; Lönnqvist, F.; Nuutila, P.; Zierath, J.R. Effects of Metformin and Rosiglitazone Treatment on Insulin Signaling and Glucose Uptake in Patients with Newly Diagnosed Type 2 Diabetes: A Randomized Controlled Study. Diabetes 2005, 54, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Shang, K.; Jia, X.; Qiao, J.; Kang, J.; Guan, Y. Endometrial Abnormality in Women with Polycystic Ovary Syndrome. Reprod. Sci. 2012, 19, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.; Rosas, C.; Kohan, K.; Gabler, F.; Vantman, D.; Romero, C.; Vega, M. Metformin Augments the Levels of Molecules That Regulate the Expression of the Insulin-Dependent Glucose Transporter GLUT4 in the Endometria of Hyperinsulinemic PCOS Patients. Hum. Reprod. 2013, 28, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Liu, C.X.; Tian, Z.R.; Jiang, Q.H.; Sun, Y.P. Effects of Metformin on the Expression of GLUT4 in Endometrium of Obese Women with Polycystic Ovary Syndrome. Biol. Reprod. 2012, 87, 1–5. [Google Scholar] [CrossRef]
- Li, X.; Cui, P.; Jiang, H.Y.; Guo, Y.R.; Pishdari, B.; Hu, M.; Feng, Y.; Billig, H.; Shao, R. Reversing the Reduced Level of Endometrial GLUT4 Expression in Polycystic Ovary Syndrome: A Mechanistic Study of Metformin Action. Am. J. Transl. Res. 2015, 7, 574–586. [Google Scholar] [PubMed]
- Zhang, L.; Liao, Q. Effects of Testosterone and Metformin on Glucose Metabolism in Endometrium. Fertil. Steril. 2010, 93, 2295–2298. [Google Scholar] [CrossRef] [PubMed]
- Świderska, E.; Strycharz, J.; Wróblewski, A.; Szemraj, J.; Drzewoski, J.; Śliwińska, A. Role of PI3K/AKT Pathway in Insulin-Mediated Glucose Uptake, Blood Glucose Levels. In Blood Glucose Levels Intech; Books on Demand: Norderstedt, Germany, 2018. [Google Scholar]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Mo, H.; Chen, W.; Li, L.; Xiao, Y.; Zhang, J.; Li, X.; Lu, Y. Role of the PI3K-Akt Signaling Pathway in the Pathogenesis of Polycystic Ovary Syndrome. Reprod. Sci. 2017, 24, 646–655. [Google Scholar] [CrossRef]
- Ujvari, D.; Hulchiy, M.; Calaby, A.; Nybacka, Å.; Byström, B.; Hirschberg, A.L. Lifestyle Intervention Up-Regulates Gene and Protein Levels of Molecules Involved in Insulin Signaling in the Endometrium of Overweight/Obese Women with Polycystic Ovary Syndrome. Hum. Reprod. 2014, 29, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Fornes, R.; Ormazabal, P.; Rosas, C.; Gabler, F.; Vantman, D.; Romero, C.; Vega, M. Changes in the Expression of Insulin Signaling Pathway Molecules in Endometria from Polycystic Ovary Syndrome Women with or without Hyperinsulinemia. Mol. Med. 2010, 16, 129–136. [Google Scholar] [CrossRef]
- Ferreira, G.D.; Germeyer, A.; de Barros Machado, A.; do Nascimento, T.L.; Strowitzki, T.; Brum, I.S.; von Eye Corleta, H.; Capp, E. Metformin Modulates PI3K and GLUT4 Expression and Akt/PKB Phosphorylation in Human Endometrial Stromal Cells after Stimulation with Androgen and Insulin. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 175, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, R.W.; Elliott, B.T. Akt/PKB Activation and Insulin Signaling: A Novel Insulin Signaling Pathway in the Treatment of Type 2 Diabetes. Diabetes. Metab. Syndr. Obes. 2014, 7, 55–64. [Google Scholar] [CrossRef]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK Signalling: Protein Kinases in Glucose Homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef]
- Kumar, N.; Dey, C.S. Metformin Enhances Insulin Signalling in Insulin-Dependent and -Independent Pathways in Insulin Resistant Muscle Cells. Br. J. Pharmacol. 2002, 137, 329–336. [Google Scholar] [CrossRef]
- Rice, S.; Pellatt, L.J.; Bryan, S.J.; Whitehead, S.A.; Mason, H.D. Action of Metformin on the Insulin-Signaling Pathway and on Glucose Transport in Human Granulosa Cells. J. Clin. Endocrinol. Metab. 2011, 96, E427–E435. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, B.; Götte, M.; Wülfing, P.; Schüring, A.N.; Kiesel, L.; Greb, R.R. Metformin Alters Insulin Signaling and Viability of Human Granulosa Cells. Fertil. Steril. 2005, 84, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Heber, M.F.; Ferreira, S.R.; Abruzzese, G.A.; Raices, T.; Pignataro, O.P.; Vega, M.; Motta, A.B. Metformin Improves Ovarian Insulin Signaling Alterations Caused by Fetal Programming. J. Endocrinol. 2019, 240, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, S.; Zhang, Z.; Lin, Q.; Liu, Y.; Xiao, Y.; Xiao, K.; Wang, Z. Defective Insulin Signaling and the Protective Effects of Dimethyldiguanide during Follicular Development in the Ovaries of Polycystic Ovary Syndrome. Mol. Med. Rep. 2017, 16, 8164–8170. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wang, B.; Tao, Y. Mitigating Effect of Metformin on Polycystic Ovarian Syndrome and Insulin Resistance in Rats, and the Mechanisms Involved. Trop. J. Pharm. Res. 2020, 19, 1941–1946. [Google Scholar] [CrossRef]
- Ma, H.M.; Chen, D.M.; Xiang, L.; Liu, C.Q.; Hou, Q.N.; He, Y.T.; Xin, C.; Zhang, Y.F.; Pei, X.Y.; Wang, Y.R.; et al. Effect of Metformin-Induced Stimulation on the Expression of Insulin Receptor Substrate 1 through Negative Regulation of P70S6k. Reprod. Dev. Med. 2018, 2, 15–20. [Google Scholar] [CrossRef]
- Kim, Y.-B.; Ciaraldi, T.P.; Kong, A.; Kim, D.; Chu, N.; Mohideen, P.; Mudaliar, S.; Henry, R.R.; Kahn, B.B. Troglitazone but Not Metformin Restores Insulin-Stimulated Phosphoinositide 3-Kinase Activity and Increases P110beta Protein Levels in Skeletal Muscle of Type 2 Diabetic Subjects. Diabetes 2002, 51, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Polianskyte-Prause, Z.; Tolvanen, T.A.; Lindfors, S.; Dumont, V.; Van, M.; Wang, H.; Dash, S.N.; Berg, M.; Naams, J.B.; Hautala, L.C.; et al. Metformin Increases Glucose Uptake and Acts Renoprotectively by Reducing SHIP2 Activity. FASEB J. 2019, 33, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Winder, W.W.; Hardie, D.G. AMP-Activated Protein Kinase, a Metabolic Master Switch: Possible Roles in Type 2 Diabetes. Am. J. Physiol. 1999, 277, E1–E10. [Google Scholar] [CrossRef] [PubMed]
- Lamoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Bailey, C.J.; Puah, J.A. Effect of Metformin on Glucose Metabolism in Mouse Soleus Muscle. Diabete Metab. 1986, 12, 212–218. [Google Scholar] [PubMed]
- Jackson, R.A.; Hawa, M.I.; Jaspan, J.B.; Sim, B.M.; Disilvio, L.; Featherbe, D.; Kurtz, A.B. Mechanism of Metformin Action in Non-Insulin-Dependent Diabetes. Diabetes 1987, 36, 632–640. [Google Scholar] [CrossRef]
- Pagano, G.; Tagliaferro, V.; Carta, Q.; Caselle, M.T.; Bozzo, C.; Vitelli, F.; Trovati, M.; Cocuzza, E. Metformin Reduces Insulin Requirement in Type 1 (Insulin-Dependent) Diabetes. Diabetologia 1983, 24, 351–354. [Google Scholar] [CrossRef]
- Pedersen, O.; Nielsen, O.H.; Bak, J.; Richelsen, B.; Beck-Nielsen, H.; Sørensen, N.S. The Effects of Metformin on Adipocyte Insulin Action and Metabolic Control in Obese Subjects with Type 2 Diabetes. Diabet. Med. 1989, 6, 249–256. [Google Scholar] [CrossRef]
- Gin, H.; Messerchmitt, C.; Brottier, E.; Aubertin, J. Metformin Improved Insulin Resistance in Type I, Insulin-Dependent, Diabetic Patients. Metabolism 1985, 34, 923–925. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Leclerc, J.; Hébrard, S.; Lantier, L.; Mounier, R.; Andreelli, F.; Foretz, M. AMP-Activated Protein Kinase in the Regulation of Hepatic Energy Metabolism: From Physiology to Therapeutic Perspectives. Acta Physiol. (Oxf.) 2009, 196, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, J.M.; Treebak, J.T.; Schjerling, P.; Goodyear, L.; Wojtaszewski, J.F.P. Two Weeks of Metformin Treatment Induces AMPK-Dependent Enhancement of Insulin-Stimulated Glucose Uptake in Mouse Soleus Muscle. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wan, Q.; Guan, Q.; Gao, L.; Zhao, J. High-Fat Diet Feeding Impairs Both the Expression and Activity of AMPKa in Rats’ Skeletal Muscle. Biochem. Biophys. Res. Commun. 2006, 339, 701–707. [Google Scholar] [CrossRef]
- Luna, V.; Casauban, L.; Sajan, M.P.; Gomez-Daspet, J.; Powe, J.L.; Miura, A.; Rivas, J.; Standaert, M.L.; Farese, R.V. Metformin Improves Atypical Protein Kinase C Activation by Insulin and Phosphatidylinositol-3,4,5-(PO4)3 in Muscle of Diabetic Subjects. Diabetologia 2006, 49, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Hirshman, M.F.; Nygren, J.; Svanfeldt, M.; Bavenholm, P.; Rooyackers, O.; Zhou, G.; Williamson, J.M.; Ljunqvist, O.; Efendic, S.; et al. Metformin Increases AMP-Activated Protein Kinase Activity in Skeletal Muscle of Subjects with Type 2 Diabetes. Diabetes 2002, 51, 2074–2081. [Google Scholar] [CrossRef]
- Sajan, M.P.; Bandyopadhyay, G.; Miura, A.; Standaert, M.L.; Nimal, S.; Longnus, S.L.; Van Obberghen, E.; Hainault, I.; Foufelle, F.; Kahn, R.; et al. AICAR and Metformin, but Not Exercise, Increase Muscle Glucose Transport through AMPK-, ERK-, and PDK1-Dependent Activation of Atypical PKC. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E179–E192. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and Molecular Mechanisms of Metformin: An Overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The Antidiabetic Drug Metformin Activates the AMP-Activated Protein Kinase Cascade via an Adenine Nucleotide-Independent Mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.-M.; Zhang, D.; Camporez, J.-P.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin Inhibits Gluconeogenesis via a Redox-Dependent Mechanism in Vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low Concentrations of Metformin Suppress Glucose Production in Hepatocytes through AMP-Activated Protein Kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Neither LKB1 nor AMPK Are the Direct Targets of Metformin. Gastroenterology 2006, 131, 973. [Google Scholar] [CrossRef] [PubMed]
- Naghiaee, Y.; Didehdar, R.; Pourrajab, F.; Rahmanian, M.; Heiranizadeh, N.; Mohiti, A.; Mohiti-Ardakani, J. Metformin Downregulates MiR223 Expression in Insulin-Resistant 3T3L1 Cells and Human Diabetic Adipose Tissue. Endocrine 2020, 70, 498–508. [Google Scholar] [CrossRef]
- Plaideau, C.; Lai, Y.-C.; Kviklyte, S.; Zanou, N.; Löfgren, L.; Andersén, H.; Vertommen, D.; Gailly, P.; Hue, L.; Bohlooly-Y, M.; et al. Effects of Pharmacological AMP Deaminase Inhibition and Ampd1 Deletion on Nucleotide Levels and AMPK Activation in Contracting Skeletal Muscle. Chem. Biol. 2014, 21, 1497–1510. [Google Scholar] [CrossRef]
- Ouyang, J.; Parakhia, R.A.; Ochs, R.S. Metformin Activates AMP Kinase through Inhibition of AMP Deaminase. J. Biol. Chem. 2011, 286, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.G.; Logan, P.J.; Jones, G.C.; Small, M.; Sattar, N.; Connell, J.M.C.; Cleland, S.J.; Salt, I.P. AMP-Activated Protein Kinase Is Activated in Adipose Tissue of Individuals with Type 2 Diabetes Treated with Metformin: A Randomised Glycaemia-Controlled Crossover Study. Diabetologia 2011, 54, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Jensterle, M.; Kravos, N.A.; Dolžan, V.; Goričar, K.; Herman, R.; Rizzo, M.; Janež, A. Glucose Transporter 4 MRNA Expression in Subcutaneous Adipose Tissue of Women with PCOS Remains Unchanged despite Metformin Withdrawal: Is There a Cellular Metabolic Treatment Legacy Effect? Endocrine 2021, 1–10. Available online: https://link.springer.com/article/10.1007%2Fs12020-021-02934-4 (accessed on 28 December 2021). [CrossRef] [PubMed]
- Holmes, B.F.; Sparling, D.P.; Olson, A.L.; Winder, W.W.; Dohm, G.L. Regulation of Muscle GLUT4 Enhancer Factor and Myocyte Enhancer Factor 2 by AMP-Activated Protein Kinase. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E1071–E1076. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: A Target for Drugs and Natural Products with Effects on Both Diabetes and Cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef]
- Lee, J.O.; Lee, S.K.; Jung, J.H.; Kim, J.H.; You, G.Y.; Kim, S.J.; Park, S.H.; Uhm, K.O.; Kim, H.S. Metformin Induces Rab4 through AMPK and Modulates GLUT4 Translocation in Skeletal Muscle Cells. J. Cell. Physiol. 2011, 226, 974–981. [Google Scholar] [CrossRef]
- Lee, J.O.; Lee, S.K.; Kim, J.H.; Kim, N.; You, G.Y.; Moon, J.W.; Kim, S.J.; Park, S.H.; Kim, H.S. Metformin Regulates Glucose Transporter 4 (GLUT4) Translocation through AMP-Activated Protein Kinase (AMPK)-Mediated Cbl/CAP Signaling in 3T3-L1 Preadipocyte Cells. J. Biol. Chem. 2012, 287, 44121–44129. [Google Scholar] [CrossRef]
- Singh, R.; Chandel, S.; Dey, D.; Ghosh, A.; Roy, S.; Ravichandiran, V.; Ghosh, D. Epigenetic Modification and Therapeutic Targets of Diabetes Mellitus. Biosci. Rep. 2020, 40, BSR20202160. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Epigenetics of Insulin Resistance: An Emerging Field in Translational Medicine. Curr. Diab. Rep. 2013, 13, 229–237. [Google Scholar] [CrossRef]
- Ling, C.; Groop, L. Epigenetics: A Molecular Link between Environmental Factors and Type 2 Diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic Effects of Metformin: From Molecular Mechanisms to Clinical Implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.-F.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin Regulates Global DNA Methylation via Mitochondrial One-Carbon Metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; van Denderen, B.J.W.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-Activated Protein Kinase Regulates GLUT4 Transcription by Phosphorylating Histone Deacetylase 5. Diabetes 2008, 57, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Milne, J.C.; Imamura, T.; Schenk, S.; Sonoda, N.; Babendure, J.L.; Lu, J.-C.; Smith, J.J.; Jirousek, M.R.; Olefsky, J.M. SIRT1 Exerts Anti-Inflammatory Effects and Improves Insulin Sensitivity in Adipocytes. Mol. Cell. Biol. 2009, 29, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Kulyté, A. MicroRNA Regulatory Networks in Human Adipose Tissue and Obesity. Nat. Rev. Endocrinol. 2015, 11, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Catellani, C.; Lazzeroni, P.; Sartori, C.; Nicoli, A.; Amarri, S.; La Sala, G.B.; Street, M.E. MiRNAs Regulating Insulin Sensitivity Are Dysregulated in Polycystic Ovary Syndrome (PCOS) Ovaries and Are Associated With Markers of Inflammation and Insulin Sensitivity. Front. Endocrinol. (Lausanne) 2019, 10, 879. [Google Scholar] [CrossRef] [PubMed]
- Esteves, J.V.; Enguita, F.J.; Machado, U.F. MicroRNAs-Mediated Regulation of Skeletal Muscle GLUT4 Expression and Translocation in Insulin Resistance. J. Diabetes Res. 2017, 2017, 7267910. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-Mediated Increase in DICER1 Regulates MicroRNA Expression and Cellular Senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef]
- Hathaway, Q.; Pinti, M.; Durr, A.; Waris, S.; Shepherd, D.; Hollander, J. Regulating MicroRNA Expression: At the Heart of Diabetes Mellitus and the Mitochondrion. Am. J. Physiol.-Hear. Circ. Physiol. 2017, 314, ajpheart.00520.2017. [Google Scholar] [CrossRef]
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin Elicits Anticancer Effects through the Sequential Modulation of DICER and C-MYC. Nat. Commun. 2012, 3, 865. [Google Scholar] [CrossRef]
- Chen, Y.H.; Heneidi, S.; Lee, J.M.; Layman, L.C.; Stepp, D.W.; Gamboa, G.M.; Chen, B.S.; Chazenbalk, G.; Azziz, R. Mirna-93 Inhibits Glut4 and Is Overexpressed in Adipose Tissue of Polycystic Ovary Syndrome Patients and Women with Insulin Resistance. Diabetes 2013, 62, 2278–2286. [Google Scholar] [CrossRef]
- Chuang, T.-Y.; Wu, H.-L.; Chen, C.-C.; Gamboa, G.M.; Layman, L.C.; Diamond, M.P.; Azziz, R.; Chen, Y.-H. MicroRNA-223 Expression Is Upregulated in Insulin Resistant Human Adipose Tissue. J. Diabetes Res. 2015, 2015, 943659. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Meng, X.; Che, H.; Shen, N.; Xiao, D.; Song, X.; Liang, M.; Fu, X.; Ju, J.; Li, Y.; et al. Regulation of Insulin Resistance by Multiple MiRNAs via Targeting the GLUT4 Signalling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 38, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Esteves, J.V.; Yonamine, C.Y.; Pinto-Junior, D.C.; Gerlinger-Romero, F.; Enguita, F.J.; Machado, U.F. Diabetes Modulates MicroRNAs 29b-3p, 29c-3p, 199a-5p and 532-3p Expression in Muscle: Possible Role in GLUT4 and HK2 Repression. Front. Endocrinol. (Lausanne) 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Mysore, R.; Ortega, F.J.; Latorre, J.; Ahonen, M.; Savolainen-Peltonen, H.; Fischer-Posovszky, P.; Wabitsch, M.; Olkkonen, V.M.; Fernández-Real, J.M.; Haridas, P.A.N. MicroRNA-221-3p Regulates Angiopoietin-Like 8 (ANGPTL8) Expression in Adipocytes. J. Clin. Endocrinol. Metab. 2017, 102, 4001–4012. [Google Scholar] [CrossRef] [PubMed]
- Massart, J.; Sjögren, R.J.O.; Lundell, L.S.; Mudry, J.M.; Franck, N.; O’Gorman, D.J.; Egan, B.; Zierath, J.R.; Krook, A. Altered MiR-29 Expression in Type 2 Diabetes Influences Glucose and Lipid Metabolism in Skeletal Muscle. Diabetes 2017, 66, 1807–1818. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Duan, L.; Wei, S.; Liu, J.; Tian, L.; Quan, J.; Zhang, Q.; Liu, J.; Yang, J. Metformin Ameliorates Skeletal Muscle Insulin Resistance by Inhibiting MiR-21 Expression in a High-Fat Dietary Rat Model. Oncotarget 2017, 8, 98029–98039. [Google Scholar] [CrossRef] [PubMed]
- Mensà, E.; Giuliani, A.; Matacchione, G.; Gurau, F.; Bonfigli, A.R.; Romagnoli, F.; De Luca, M.; Sabbatinelli, J.; Olivieri, F. Circulating MiR-146a in Healthy Aging and Type 2 Diabetes: Age- and Gender-Specific Trajectories. Mech. Ageing Dev. 2019, 180. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; Enlund, E.; Funcke, J.-B.; Tews, D.; Holzmann, K.; Debatin, K.-M.; Wabitsch, M.; Fischer-Posovszky, P. MiR-146a-Mediated Suppression of the Inflammatory Response in Human Adipocytes. Sci. Rep. 2016, 6, 38339. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanyam, M.; Aravind, S.; Gokulakrishnan, K.; Prabu, P.; Sathishkumar, C.; Ranjani, H.; Mohan, V. Impaired MiR-146a Expression Links Subclinical Inflammation and Insulin Resistance in Type 2 Diabetes. Mol. Cell. Biochem. 2011, 351, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Mercader, J.M.; Moreno-Navarrete, J.M.; Rovira, O.; Guerra, E.; Esteve, E.; Xifra, G.; Martínez, C.; Ricart, W.; Rieusset, J.; et al. Profiling of Circulating MicroRNAs Reveals Common MicroRNAs Linked to Type 2 Diabetes That Change with Insulin Sensitization. Diabetes Care 2014, 37, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Ghai, V.; Kim, T.-K.; Etheridge, A.; Nielsen, T.; Hansen, T.; Pedersen, O.; Galas, D.; Wang, K. Extracellular Vesicle Encapsulated MicroRNAs in Patients with Type 2 Diabetes Are Affected by Metformin Treatment. J. Clin. Med. 2019, 8, 617. [Google Scholar] [CrossRef] [PubMed]
- Udesen, P.B.; Glintborg, D.; Sørensen, A.E.; Svendsen, R.; Nielsen, N.L.S.; Wissing, M.L.M.; Andersen, M.S.; Englund, A.L.M.; Dalgaard, L.T. Metformin Decreases MiR-122, MiR-223 and MiR-29a in Women with Polycystic Ovary Syndrome. Endocr. Connect. 2020, 9, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- García-Calzón, S.; Perfilyev, A.; Martinell, M.; Ustinova, M.; Kalamajski, S.; Franks, P.W.; Bacos, K.; Elbere, I.; Pihlajamäki, J.; Volkov, P.; et al. Epigenetic Markers Associated with Metformin Response and Intolerance in Drug-Naïve Patients with Type 2 Diabetes. Sci. Transl. Med. 2020, 12, 561. [Google Scholar] [CrossRef]
- Takahashi, N.; Kimura, A.P.; Otsuka, K.; Ohmura, K.; Naito, S.; Yoshida, M.; Ieko, M. Dreh, a Long Noncoding RNA Repressed by Metformin, Regulates Glucose Transport in C2C12 Skeletal Muscle Cells. Life Sci. 2019, 236, 116906. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Tong, L.; Song, Y.; Sun, J.; Shi, J.; Wu, Z.; Diao, Y.; Li, Y.; Wang, Z. Long Noncoding RNA H19 Participates in Metformin-Mediated Inhibition of Gastric Cancer Cell Invasion. J. Cell. Physiol. 2019, 234, 4515–4527. [Google Scholar] [CrossRef] [PubMed]
- Gui, W.; Zhu, W.F.; Zhu, Y.; Tang, S.; Zheng, F.; Yin, X.; Lin, X.; Li, H. LncRNAH19 Improves Insulin Resistance in Skeletal Muscle by Regulating Heterogeneous Nuclear Ribonucleoprotein A1. Cell Commun. Signal. 2020, 18, 173. [Google Scholar] [CrossRef]
- Mueckler, M. Insulin Resistance and the Disruption of Glut4 Trafficking in Skeletal Muscle. J. Clin. Investig. 2001, 107, 1211–1213. [Google Scholar] [CrossRef]
- Shepherd, P.R.; Kahn, B.B. Glucose Transporters and Insulin Action--Implications for Insulin Resistance and Diabetes Mellitus. N. Engl. J. Med. 1999, 341, 248–257. [Google Scholar] [CrossRef]
- Xu, P.-T.; Song, Z.; Zhang, W.-C.; Jiao, B.; Yu, Z.-B. Impaired Translocation of GLUT4 Results in Insulin Resistance of Atrophic Soleus Muscle. Biomed Res. Int. 2015, 2015, 291987. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Lavigne, C.; Jacques, H.; Marette, A. Defective Insulin-Induced GLUT4 Translocation in Skeletal Muscle of High Fat-Fed Rats Is Associated with Alterations in Both Akt/Protein Kinase B and Atypical Protein Kinase C (Zeta/Lambda) Activities. Diabetes 2001, 50, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, L.J.; Kahn, B.B. Exercise, Glucose Transport, and Insulin Sensitivity. Annu. Rev. Med. 1998, 49, 235–261. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, P.R.; Withers, D.J.; Siddle, K. Phosphoinositide 3-Kinase: The Key Switch Mechanism in Insulin Signalling. Biochem. J. 1998, 333, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Purintrapiban, J.; Ratanachaiyavong, S.; Mcshane, H. The Effects of Insulin and Metformin on Glucose Uptake in L8 Myotubes. ScienceAsia 2003, 29, 341. [Google Scholar] [CrossRef]
- Watson, R.T.; Pessin, J.E. Bridging the GAP between Insulin Signaling and GLUT4 Translocation. Trends Biochem. Sci. 2006, 31, 215–222. [Google Scholar] [CrossRef]
- Gao, L.; Chen, J.; Gao, J.; Wang, H.; Xiong, W. Super-Resolution Microscopy Reveals the Insulin-Resistance-Regulated Reorganization of GLUT4 on Plasma Membranes. J. Cell Sci. 2017, 130, 396–405. [Google Scholar] [CrossRef]
- Corrêa-Giannella, M.L.; Machado, U.F. SLC2A4gene: A Promising Target for Pharmacogenomics of Insulin Resistance. Pharmacogenomics 2013, 14, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Fischer, Y.; Thomas, J.; Rösen, P.; Kammermeier, H. Action of Metformin on Glucose Transport and Glucose Transporter Glut1 and Glut4 in Heart Muscle Cells from Healthy and Diabetic Rats. Endocrinology 1995, 136, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, S.; Hamann, A.; Klein, H.H.; Benecke, H.; Kreymann, G.; Flier, J.S.; Greten, H. Association of Metformin’s Effect to Increase Insulin-Stimulated Glucose Transport with Potentiation of Insulin-Induced Translocation of Glucose Transporters from Intracellular Pool to Plasma Membrane in Rat Adipocytes. Diabetes 1991, 40, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Sarabia, V.; Lam, L.; Burdett, E.; Leiter, L.A.; Klip, A. Glucose Transport in Human Skeletal Muscle Cells in Culture. Stimulation by Insulin and Metformin. J. Clin. Investig. 1992, 90, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Holman, G.D. Long-Term Metformin Treatment Stimulates Cardiomyocyte Glucose Transport through an AMP-Activated Protein Kinase-Dependent Reduction in GLUT4 Endocytosis. Endocrinology 2006, 147, 2728–2736. [Google Scholar] [CrossRef] [PubMed]
- Detaille, D.; Wiernsperger, N.; Devos, P. Metformin Interaction with Insulin-Regulated Glucose Uptake, Using the Xenopus Laevis Oocyte Model Expressing the Mammalian Transporter GLUT4. Eur. J. Pharmacol. 1999, 377, 127–136. [Google Scholar] [CrossRef]
- Matthaei, S.; Reibold, J.P.; Hamann, A.; Benecke, H.; Häring, H.U.; Greten, H.; Klein, H.H. In Vivo Metformin Treatment Ameliorates Insulin Resistance: Evidence for Potentiation of Insulin-Induced Translocation and Increased Functional Activity of Glucose Transporters in Obese (Fa/Fa) Zucker Rat Adipocytes. Endocrinology 1993, 133, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Baus, D.; Heermeier, K.; De Hoop, M.; Metz-Weidmann, C.; Gassenhuber, J.; Dittrich, W.; Welte, S.; Tennagels, N. Identification of a Novel AS160 Splice Variant That Regulates GLUT4 Translocation and Glucose-Uptake in Rat Muscle Cells. Cell. Signal. 2008, 20, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-Stimulated Phosphorylation of a Rab GTPase-Activating Protein Regulates GLUT4 Translocation. J. Biol. Chem. 2003, 278, 14599–14602. [Google Scholar] [CrossRef] [PubMed]
References
- Klip, A.; McGraw, T.E.; James, D.E. Thirty Sweet Years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381.
- Nolan, C.J.; Prentki, M. Insulin Resistance and Insulin Hypersecretion in the Metabolic Syndrome and Type 2 Diabetes: Time for a Conceptual Framework Shift. Diabetes Vasc. Dis. Res. 2019, 16, 118–127.
- Thorens, B.; Mueckler, M. Glucose Transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145.
- James, D.E.; Brown, R.; Navarro, J.; Pilch, P.F. Insulin-Regulatable Tissues Express a Unique Insulin-Sensitive Glucose Transport Protein. Nature 1988, 333, 183–185.
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted Disruption of the Glucose Transporter 4 Selectively in Muscle Causes Insulin Resistance and Glucose Intolerance. Nat. Med. 2000, 6, 924–928.
- Cline, G.W.; Petersen, K.F.; Krssak, M.; Shen, J.; Hundal, R.S.; Trajanoski, Z.; Inzucchi, S.; Dresner, A.; Rothman, D.L.; Shulman, G.I. Impaired Glucose Transport as a Cause of Decreased Insulin-Stimulated Muscle Glycogen Synthesis in Type 2 Diabetes. N. Engl. J. Med. 1999, 341, 240–246.
- Armoni, M.; Harel, C.; Karnieli, E. GLUT4 Goes Abnormal: Disregulation of the Insulin-Responsive Glucose Transporter in Abnormal Metabolic States. Curr. Med. Chem.-Immunol. Endocr. Metab. Agents 2005, 5, 207–217.
- Karnieli, E.; Zarnowski, M.J.; Hissin, P.J.; Simpson, I.A.; Salans, L.B.; Cushman, S.W. Insulin-Stimulated Translocation of Glucose Transport Systems in the Isolated Rat Adipose Cell. Time Course, Reversal, Insulin Concentration Dependency, and Relationship to Glucose Transport Activity. J. Biol. Chem. 1981, 256, 4772–4777.
- Karnieli, E.; Barzilai, A.; Rafaeloff, R.; Armoni, M. Distribution of Glucose Transporters in Membrane Fractions Isolated from Human Adipose Cells. Relation to Cell Size. J. Clin. Investig. 1986, 78, 1051–1055.
- Leguisamo, N.M.; Lehnen, A.M.; Machado, U.F.; Okamoto, M.M.; Markoski, M.M.; Pinto, G.H.; Schaan, B.D. GLUT4 Content Decreases along with Insulin Resistance and High Levels of Inflammatory Markers in Rats with Metabolic Syndrome. Cardiovasc. Diabetol. 2012, 11, 100.
- Seböková, E.; Klimes, I.; Moss, R.; Mitková, A.; Wiersma, M.; Bohov, P. Decreased Glucose Transporter Protein (GLUT4) in Skeletal Muscle of Hypertriglyceridaemic Insulin-Resistant Rat. Physiol. Res. 1995, 44, 87–92.
- Napoli, R.; Hirshman, M.F.; Horton, E.S. Mechanisms and Time Course of Impaired Skeletal Muscle Glucose Transport Activity in Streptozocin Diabetic Rats. J. Clin. Investig. 1995, 96, 427–437.
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-Selective Targeting of the GLUT4 Gene Impairs Insulin Action in Muscle and Liver. Nature 2001, 409, 729–733.
- Graham, T.E.; Kahn, B.B. Tissue-Specific Alterations of Glucose Transport and Molecular Mechanisms of Intertissue Communication in Obesity and Type 2 Diabetes. Horm. Metab. Res. Horm. Stoffwechselforsch. Horm. Metab. 2007, 39, 717–721.
- Rossetti, L.; Stenbit, A.E.; Chen, W.; Hu, M.; Barzilai, N.; Katz, E.B.; Charron, M.J. Peripheral but Not Hepatic Insulin Resistance in Mice with One Disrupted Allele of the Glucose Transporter Type 4 (GLUT4) Gene. J. Clin. Investig. 1997, 100, 1831–1839.
- Stenbit, A.E.; Tsao, T.S.; Li, J.; Burcelin, R.; Geenen, D.L.; Factor, S.M.; Houseknecht, K.; Katz, E.B.; Charron, M.J. GLUT4 Heterozygous Knockout Mice Develop Muscle Insulin Resistance and Diabetes. Nat. Med. 1997, 3, 1096–1101.
- Li, J.; Houseknecht, K.L.; Stenbit, A.E.; Katz, E.B.; Charron, M.J. Reduced Glucose Uptake Precedes Insulin Signaling Defects in Adipocytes from Heterozygous GLUT4 Knockout Mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 1117–1125.
- Shepherd, P.R.; Gnudi, L.; Tozzo, E.; Yang, H.; Leach, F.; Kahn, B.B. Adipose Cell Hyperplasia and Enhanced Glucose Disposal in Transgenic Mice Overexpressing GLUT4 Selectively in Adipose Tissue. J. Biol. Chem. 1993, 268, 22243–22246.
- Carvalho, E.; Kotani, K.; Peroni, O.D.; Kahn, B.B. Adipose-Specific Overexpression of GLUT4 Reverses Insulin Resistance and Diabetes in Mice Lacking GLUT4 Selectively in Muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E551–E561.
- Brozinick, J.T.J.; McCoid, S.C.; Reynolds, T.H.; Nardone, N.A.; Hargrove, D.M.; Stevenson, R.W.; Cushman, S.W.; Gibbs, E.M. GLUT4 Overexpression in Db/Db Mice Dose-Dependently Ameliorates Diabetes but Is Not a Lifelong Cure. Diabetes 2001, 50, 593–600.
- Tsao, T.S.; Burcelin, R.; Katz, E.B.; Huang, L.; Charron, M.J. Enhanced Insulin Action Due to Targeted GLUT4 Overexpression Exclusively in Muscle. Diabetes 1996, 45, 28–36.
- Atkinson, B.J.; Griesel, B.A.; King, C.D.; Josey, M.A.; Olson, A.L. Moderate GLUT4 Overexpression Improves Insulin Sensitivity and Fasting Triglyceridemia in High-Fat Diet-Fed Transgenic Mice. Diabetes 2013, 62, 2249–2258.
- Garvey, W.T.; Maianu, L.; Huecksteadt, T.P.; Birnbaum, M.J.; Molina, J.M.; Ciaraldi, T.P. Pretranslational Suppression of a Glucose Transporter Protein Causes Insulin Resistance in Adipocytes from Patients with Non-Insulin-Dependent Diabetes Mellitus and Obesity. J. Clin. Investig. 1991, 87, 1072–1081.
- Rosenbaum, D.; Haber, R.S.; Dunaif, A. Insulin Resistance in Polycystic Ovary Syndrome: Decreased Expression of GLUT-4 Glucose Transporters in Adipocytes. Am. J. Physiol.-Endocrinol. Metab. 1993, 264, E161–E166.
- Garvey, W.T.; Maianu, L.; Zhu, J.H.; Brechtel-Hook, G.; Wallace, P.; Baron, A.D. Evidence for Defects in the Trafficking and Translocation of GLUT4 Glucose Transporters in Skeletal Muscle as a Cause of Human Insulin Resistance. J. Clin. Investig. 1998, 101, 2377–2386.
- Kampmann, U.; Christensen, B.; Nielsen, T.S.; Pedersen, S.B.; Ørskov, L.; Lund, S.; Møller, N.; Jessen, N. GLUT4 and UBC9 Protein Expression Is Reduced in Muscle from Type 2 Diabetic Patients with Severe Insulin Resistance. PLoS ONE 2011, 6, e27854.
- Bunprajun, T.; Henriksen, T.I.; Scheele, C.; Pedersen, B.K.; Green, C.J. Lifelong Physical Activity Prevents Aging-Associated Insulin Resistance in Human Skeletal Muscle Myotubes via Increased Glucose Transporter Expression. PLoS ONE 2013, 8, e66628.
- Biensø, R.S.; Ringholm, S.; Kiilerich, K.; Aachmann-Andersen, N.-J.; Krogh-Madsen, R.; Guerra, B.; Plomgaard, P.; van Hall, G.; Treebak, J.T.; Saltin, B.; et al. GLUT4 and Glycogen Synthase Are Key Players in Bed Rest-Induced Insulin Resistance. Diabetes 2012, 61, 1090–1099.
- Ezeh, U.; Chen, I.Y.-D.; Chen, Y.-H.; Azziz, R. Adipocyte Insulin Resistance in PCOS: Relationship With GLUT-4 Expression and Whole-Body Glucose Disposal and β-Cell Function. J. Clin. Endocrinol. Metab. 2020, 105, e2408–e2420.
- Ezeh, U.; Chen, I.Y.-D.; Chen, Y.-H.; Azziz, R. Adipocyte Expression of Glucose Transporter 1 and 4 in PCOS: Relationship to Insulin-Mediated and Non-Insulin-Mediated Whole-Body Glucose Uptake. Clin. Endocrinol. (Oxf.) 2019, 90, 542–552.
- Yan, Y.; Kover, K.L.; Moore, W.V. New Insight into Metformin Mechanism of Action and Clinical Application. In Metformin; IntechOpen: London, UK, 2020.
- Giannarelli, R.; Aragona, M.; Coppelli, A.; Del Prato, S. Reducing Insulin Resistance with Metformin: The Evidence Today. Diabetes Metab. 2003, 29, 6S28–6S35.
- Pala, L.; Barbaro, V.; Dicembrini, I.; Rotella, C.M. The Therapy of Insulin Resistance in Other Diseases besides Type 2 Diabetes. Eat. Weight Disord. 2014, 19, 275–283.
- Kamenova, P. Therapeutic Potential of Metformin in Normal Glucose Tolerant Persons with Metabolic Syndrome. Biotechnol. Biotechnol. Equip. 2020, 34, 30–37.
- Malin, S.K.; Gerber, R.; Chipkin, S.R.; Braun, B. Independent and Combined Effects of Exercise Training and Metformin on Insulin Sensitivity in Individuals with Prediabetes. Diabetes Care 2012, 35, 131–136.
- Worsley, R.; Jane, F.; Robinson, P.J.; Bell, R.J.; Davis, S.R. Metformin for Overweight Women at Midlife: A Double-Blind, Randomized, Controlled Trial. Climacteric 2015, 18, 270–277.
- Nawrocka-Rutkowska, J.; Ciecwiez, S.; Marciniak, A.; Brodowska, A.; Wiśniewska, B.; Kotlega, D.; Starczewski, A. Insulin Resistance Assessment in Patients with Polycystic Ovary Syndrome Using Different Diagnostic Criteria–Impact of Metformin Treatment. Ann. Agric. Environ. Med. 2013, 20, 528–532.
- Bjornstad, P.; Schäfer, M.; Truong, U.; Cree-Green, M.; Pyle, L.; Baumgartner, A.; Garcia Reyes, Y.; Maniatis, A.; Nayak, S.; Wadwa, R.P.; et al. Metformin Improves Insulin Sensitivity and Vascular Health in Youth With Type 1 Diabetes Mellitus. Circulation 2018, 138, 2895–2907.
- Ghaffari, M.; Payami, S.; Payami, S.; Ashtary-Larky, D.; Nikzamir, A.; Mohammadzadeh, G. Evaluation of Insulin Resistance Indices in Type 2 Diabetic Patients Treated with Different Anti-Diabetic Drugs. Open J. Endocr. Metab. Dis. 2016, 6, 95–101.
- Agarwal, S.M.; Panda, R.; Costa-Dookhan, K.A.; MacKenzie, N.E.; Treen, Q.C.; Caravaggio, F.; Hashim, E.; Leung, G.; Kirpalani, A.; Matheson, K.; et al. Metformin for Early Comorbid Glucose Dysregulation and Schizophrenia Spectrum Disorders: A Pilot Double-Blind Randomized Clinical Trial. Transl. Psychiatry 2021, 11, 219.
- Atabek, M.E.; Pirgon, O. Use of Metformin in Obese Adolescents with Hyperinsulinemia: A 6-Month, Randomized, Double-Blind, Placebo-Controlled Clinical Trial. J. Pediatr. Endocrinol. Metab. 2008, 21, 339–348.
- Fux Otta, C.; Wior, M.; Iraci, G.S.; Kaplan, R.; Torres, D.; Gaido, M.I.; Wyse, E.P. Clinical, Metabolic, and Endocrine Parameters in Response to Metformin and Lifestyle Intervention in Women with Polycystic Ovary Syndrome: A Randomized, Double-Blind, and Placebo Control Trial. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2010, 26, 173–178.
- Brufani, C.; Crinò, A.; Fintini, D.; Patera, P.I.; Cappa, M.; Manco, M. Systematic Review of Metformin Use in Obese Nondiabetic Children and Adolescents. Horm. Res. Paediatr. 2013, 80, 78–85.
- Wróbel, M.P.; Marek, B.; Kajdaniuk, D.; Rokicka, D.; Szymborska-Kajanek, A.; Strojek, K. Metformin-A New Old Drug. Endokrynol. Pol. 2017, 68, 482–496.
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. (Lausanne) 2020, 11, 191.
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the Gastrointestinal Tract. Diabetologia 2016, 59, 426–435.
- Zhang, Q.; Hu, N. Effects of Metformin on the Gut Microbiota in Obesity and Type 2 Diabetes Mellitus. Diabetes. Metab. Syndr. Obes. 2020, 13, 5003–5014.
- Kozka, I.J.; Holman, G.D. Metformin Blocks Downregulation of Cell Surface GLUT4 Caused by Chronic Insulin Treatment of Rat Adipocytes. Diabetes 1993, 42, 1159–1165.
- Pryor, P.R.; Liu, S.C.; Clark, A.E.; Yang, J.; Holman, G.D.; Tosh, D. Chronic Insulin Effects on Insulin Signalling and GLUT4 Endocytosis Are Reversed by Metformin. Biochem. J. 2000, 348, 83–91.
- Zayed, E.A.; AinShoka, A.A.; El Shazly, K.A.; Abd El Latif, H.A. Improvement of Insulin Resistance via Increase of GLUT4 and PPARγ in Metabolic Syndrome-Induced Rats Treated with Omega-3 Fatty Acid or l-Carnitine. J. Biochem. Mol. Toxicol. 2018, 32, e22218.
- Shen, X.; Wang, L.; Zhou, N.; Gai, S.; Liu, X.; Zhang, S. Beneficial Effects of Combination Therapy of Phloretin and Metformin in Streptozotocin-Induced Diabetic Rats and Improved Insulin Sensitivity in Vitro. Food Funct. 2020, 11, 392–403.
- Grisouard, J.; Timper, K.; Radimerski, T.M.; Frey, D.M.; Peterli, R.; Kola, B.; Korbonits, M.; Herrmann, P.; Krähenbühl, S.; Zulewski, H.; et al. Mechanisms of Metformin Action on Glucose Transport and Metabolism in Human Adipocytes. Biochem. Pharmacol. 2010, 80, 1736–1745.
- Al-Khalili, L.; Forsgren, M.; Kannisto, K.; Zierath, J.R.; Lönnqvist, F.; Krook, A. Enhanced Insulin-Stimulated Glycogen Synthesis in Response to Insulin, Metformin or Rosiglitazone Is Associated with Increased MRNA Expression of GLUT4 and Peroxisomal Proliferator Activator Receptor Gamma Co-Activator 1. Diabetologia 2005, 48, 1173–1179.
- Jensterle, M.; Janez, A.; Mlinar, B.; Marc, J.; Prezelj, J.; Pfeifer, M. Impact of Metformin and Rosiglitazone Treatment on Glucose Transporter 4 MRNA Expression in Women with Polycystic Ovary Syndrome. Eur. J. Endocrinol. 2008, 158, 793–801.
- Ciaraldi, T.P.; Kong, A.P.S.; Chu, N.V.; Kim, D.D.; Baxi, S.; Loviscach, M.; Plodkowski, R.; Reitz, R.; Caulfield, M.; Mudaliar, S.; et al. Regulation of Glucose Transport and Insulin Signaling by Troglitazone or Metformin in Adipose Tissue of Type 2 Diabetic Subjects. Diabetes 2002, 51, 30–36.
- Karlsson, H.K.R.; Hällsten, K.; Björnholm, M.; Tsuchida, H.; Chibalin, A.V.; Virtanen, K.A.; Heinonen, O.J.; Lönnqvist, F.; Nuutila, P.; Zierath, J.R. Effects of Metformin and Rosiglitazone Treatment on Insulin Signaling and Glucose Uptake in Patients with Newly Diagnosed Type 2 Diabetes: A Randomized Controlled Study. Diabetes 2005, 54, 1459–1467.
- Shang, K.; Jia, X.; Qiao, J.; Kang, J.; Guan, Y. Endometrial Abnormality in Women with Polycystic Ovary Syndrome. Reprod. Sci. 2012, 19, 674–683.
- Carvajal, R.; Rosas, C.; Kohan, K.; Gabler, F.; Vantman, D.; Romero, C.; Vega, M. Metformin Augments the Levels of Molecules That Regulate the Expression of the Insulin-Dependent Glucose Transporter GLUT4 in the Endometria of Hyperinsulinemic PCOS Patients. Hum. Reprod. 2013, 28, 2235–2244.
- Zhai, J.; Liu, C.X.; Tian, Z.R.; Jiang, Q.H.; Sun, Y.P. Effects of Metformin on the Expression of GLUT4 in Endometrium of Obese Women with Polycystic Ovary Syndrome. Biol. Reprod. 2012, 87, 1–5.
- Li, X.; Cui, P.; Jiang, H.Y.; Guo, Y.R.; Pishdari, B.; Hu, M.; Feng, Y.; Billig, H.; Shao, R. Reversing the Reduced Level of Endometrial GLUT4 Expression in Polycystic Ovary Syndrome: A Mechanistic Study of Metformin Action. Am. J. Transl. Res. 2015, 7, 574–586.
- Zhang, L.; Liao, Q. Effects of Testosterone and Metformin on Glucose Metabolism in Endometrium. Fertil. Steril. 2010, 93, 2295–2298.
- Świderska, E.; Strycharz, J.; Wróblewski, A.; Szemraj, J.; Drzewoski, J.; Śliwińska, A. Role of PI3K/AKT Pathway in Insulin-Mediated Glucose Uptake, Blood Glucose Levels. In Blood Glucose Levels Intech; Books on Demand: Norderstedt, Germany, 2018.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J. Clin. Investig. 2001, 108, 1167–1174.
- Li, T.; Mo, H.; Chen, W.; Li, L.; Xiao, Y.; Zhang, J.; Li, X.; Lu, Y. Role of the PI3K-Akt Signaling Pathway in the Pathogenesis of Polycystic Ovary Syndrome. Reprod. Sci. 2017, 24, 646–655.
- Ujvari, D.; Hulchiy, M.; Calaby, A.; Nybacka, Å.; Byström, B.; Hirschberg, A.L. Lifestyle Intervention Up-Regulates Gene and Protein Levels of Molecules Involved in Insulin Signaling in the Endometrium of Overweight/Obese Women with Polycystic Ovary Syndrome. Hum. Reprod. 2014, 29, 1526–1535.
- Fornes, R.; Ormazabal, P.; Rosas, C.; Gabler, F.; Vantman, D.; Romero, C.; Vega, M. Changes in the Expression of Insulin Signaling Pathway Molecules in Endometria from Polycystic Ovary Syndrome Women with or without Hyperinsulinemia. Mol. Med. 2010, 16, 129–136.
- Ferreira, G.D.; Germeyer, A.; de Barros Machado, A.; do Nascimento, T.L.; Strowitzki, T.; Brum, I.S.; von Eye Corleta, H.; Capp, E. Metformin Modulates PI3K and GLUT4 Expression and Akt/PKB Phosphorylation in Human Endometrial Stromal Cells after Stimulation with Androgen and Insulin. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 175, 157–162.
- Mackenzie, R.W.; Elliott, B.T. Akt/PKB Activation and Insulin Signaling: A Novel Insulin Signaling Pathway in the Treatment of Type 2 Diabetes. Diabetes. Metab. Syndr. Obes. 2014, 7, 55–64.
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK Signalling: Protein Kinases in Glucose Homeostasis. Expert Rev. Mol. Med. 2012, 14, e1.
- Kumar, N.; Dey, C.S. Metformin Enhances Insulin Signalling in Insulin-Dependent and -Independent Pathways in Insulin Resistant Muscle Cells. Br. J. Pharmacol. 2002, 137, 329–336.
- Rice, S.; Pellatt, L.J.; Bryan, S.J.; Whitehead, S.A.; Mason, H.D. Action of Metformin on the Insulin-Signaling Pathway and on Glucose Transport in Human Granulosa Cells. J. Clin. Endocrinol. Metab. 2011, 96, E427–E435.
- Sonntag, B.; Götte, M.; Wülfing, P.; Schüring, A.N.; Kiesel, L.; Greb, R.R. Metformin Alters Insulin Signaling and Viability of Human Granulosa Cells. Fertil. Steril. 2005, 84, 1173–1179.
- Heber, M.F.; Ferreira, S.R.; Abruzzese, G.A.; Raices, T.; Pignataro, O.P.; Vega, M.; Motta, A.B. Metformin Improves Ovarian Insulin Signaling Alterations Caused by Fetal Programming. J. Endocrinol. 2019, 240, 431–443.
- Wang, F.; Wang, S.; Zhang, Z.; Lin, Q.; Liu, Y.; Xiao, Y.; Xiao, K.; Wang, Z. Defective Insulin Signaling and the Protective Effects of Dimethyldiguanide during Follicular Development in the Ovaries of Polycystic Ovary Syndrome. Mol. Med. Rep. 2017, 16, 8164–8170.
- Hu, L.; Wang, B.; Tao, Y. Mitigating Effect of Metformin on Polycystic Ovarian Syndrome and Insulin Resistance in Rats, and the Mechanisms Involved. Trop. J. Pharm. Res. 2020, 19, 1941–1946.
- Ma, H.M.; Chen, D.M.; Xiang, L.; Liu, C.Q.; Hou, Q.N.; He, Y.T.; Xin, C.; Zhang, Y.F.; Pei, X.Y.; Wang, Y.R.; et al. Effect of Metformin-Induced Stimulation on the Expression of Insulin Receptor Substrate 1 through Negative Regulation of P70S6k. Reprod. Dev. Med. 2018, 2, 15–20.
- Kim, Y.-B.; Ciaraldi, T.P.; Kong, A.; Kim, D.; Chu, N.; Mohideen, P.; Mudaliar, S.; Henry, R.R.; Kahn, B.B. Troglitazone but Not Metformin Restores Insulin-Stimulated Phosphoinositide 3-Kinase Activity and Increases P110beta Protein Levels in Skeletal Muscle of Type 2 Diabetic Subjects. Diabetes 2002, 51, 443–448.
- Polianskyte-Prause, Z.; Tolvanen, T.A.; Lindfors, S.; Dumont, V.; Van, M.; Wang, H.; Dash, S.N.; Berg, M.; Naams, J.B.; Hautala, L.C.; et al. Metformin Increases Glucose Uptake and Acts Renoprotectively by Reducing SHIP2 Activity. FASEB J. 2019, 33, 2858–2869.
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135.
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800.
- Winder, W.W.; Hardie, D.G. AMP-Activated Protein Kinase, a Metabolic Master Switch: Possible Roles in Type 2 Diabetes. Am. J. Physiol. 1999, 277, E1–E10.
- Lamoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96.
- Bailey, C.J.; Puah, J.A. Effect of Metformin on Glucose Metabolism in Mouse Soleus Muscle. Diabete Metab. 1986, 12, 212–218.
- Jackson, R.A.; Hawa, M.I.; Jaspan, J.B.; Sim, B.M.; Disilvio, L.; Featherbe, D.; Kurtz, A.B. Mechanism of Metformin Action in Non-Insulin-Dependent Diabetes. Diabetes 1987, 36, 632–640.
- Pagano, G.; Tagliaferro, V.; Carta, Q.; Caselle, M.T.; Bozzo, C.; Vitelli, F.; Trovati, M.; Cocuzza, E. Metformin Reduces Insulin Requirement in Type 1 (Insulin-Dependent) Diabetes. Diabetologia 1983, 24, 351–354.
- Pedersen, O.; Nielsen, O.H.; Bak, J.; Richelsen, B.; Beck-Nielsen, H.; Sørensen, N.S. The Effects of Metformin on Adipocyte Insulin Action and Metabolic Control in Obese Subjects with Type 2 Diabetes. Diabet. Med. 1989, 6, 249–256.
- Gin, H.; Messerchmitt, C.; Brottier, E.; Aubertin, J. Metformin Improved Insulin Resistance in Type I, Insulin-Dependent, Diabetic Patients. Metabolism 1985, 34, 923–925.
- Viollet, B.; Guigas, B.; Leclerc, J.; Hébrard, S.; Lantier, L.; Mounier, R.; Andreelli, F.; Foretz, M. AMP-Activated Protein Kinase in the Regulation of Hepatic Energy Metabolism: From Physiology to Therapeutic Perspectives. Acta Physiol. (Oxf.) 2009, 196, 81–98.
- Kristensen, J.M.; Treebak, J.T.; Schjerling, P.; Goodyear, L.; Wojtaszewski, J.F.P. Two Weeks of Metformin Treatment Induces AMPK-Dependent Enhancement of Insulin-Stimulated Glucose Uptake in Mouse Soleus Muscle. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, 1099–1109.
- Liu, Y.; Wan, Q.; Guan, Q.; Gao, L.; Zhao, J. High-Fat Diet Feeding Impairs Both the Expression and Activity of AMPKa in Rats’ Skeletal Muscle. Biochem. Biophys. Res. Commun. 2006, 339, 701–707.
- Luna, V.; Casauban, L.; Sajan, M.P.; Gomez-Daspet, J.; Powe, J.L.; Miura, A.; Rivas, J.; Standaert, M.L.; Farese, R.V. Metformin Improves Atypical Protein Kinase C Activation by Insulin and Phosphatidylinositol-3,4,5-(PO4)3 in Muscle of Diabetic Subjects. Diabetologia 2006, 49, 375–382.
- Musi, N.; Hirshman, M.F.; Nygren, J.; Svanfeldt, M.; Bavenholm, P.; Rooyackers, O.; Zhou, G.; Williamson, J.M.; Ljunqvist, O.; Efendic, S.; et al. Metformin Increases AMP-Activated Protein Kinase Activity in Skeletal Muscle of Subjects with Type 2 Diabetes. Diabetes 2002, 51, 2074–2081.
- Sajan, M.P.; Bandyopadhyay, G.; Miura, A.; Standaert, M.L.; Nimal, S.; Longnus, S.L.; Van Obberghen, E.; Hainault, I.; Foufelle, F.; Kahn, R.; et al. AICAR and Metformin, but Not Exercise, Increase Muscle Glucose Transport through AMPK-, ERK-, and PDK1-Dependent Activation of Atypical PKC. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E179–E192.
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and Molecular Mechanisms of Metformin: An Overview. Clin. Sci. 2012, 122, 253–270.
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The Antidiabetic Drug Metformin Activates the AMP-Activated Protein Kinase Cascade via an Adenine Nucleotide-Independent Mechanism. Diabetes 2002, 51, 2420–2425.
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.-M.; Zhang, D.; Camporez, J.-P.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin Inhibits Gluconeogenesis via a Redox-Dependent Mechanism in Vivo. Nat. Med. 2018, 24, 1384–1394.
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low Concentrations of Metformin Suppress Glucose Production in Hepatocytes through AMP-Activated Protein Kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446.
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1646.
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008.
- Hardie, D.G. Neither LKB1 nor AMPK Are the Direct Targets of Metformin. Gastroenterology 2006, 131, 973.
- Naghiaee, Y.; Didehdar, R.; Pourrajab, F.; Rahmanian, M.; Heiranizadeh, N.; Mohiti, A.; Mohiti-Ardakani, J. Metformin Downregulates MiR223 Expression in Insulin-Resistant 3T3L1 Cells and Human Diabetic Adipose Tissue. Endocrine 2020, 70, 498–508.
- Plaideau, C.; Lai, Y.-C.; Kviklyte, S.; Zanou, N.; Löfgren, L.; Andersén, H.; Vertommen, D.; Gailly, P.; Hue, L.; Bohlooly-Y, M.; et al. Effects of Pharmacological AMP Deaminase Inhibition and Ampd1 Deletion on Nucleotide Levels and AMPK Activation in Contracting Skeletal Muscle. Chem. Biol. 2014, 21, 1497–1510.
- Ouyang, J.; Parakhia, R.A.; Ochs, R.S. Metformin Activates AMP Kinase through Inhibition of AMP Deaminase. J. Biol. Chem. 2011, 286, 1–11.
- Boyle, J.G.; Logan, P.J.; Jones, G.C.; Small, M.; Sattar, N.; Connell, J.M.C.; Cleland, S.J.; Salt, I.P. AMP-Activated Protein Kinase Is Activated in Adipose Tissue of Individuals with Type 2 Diabetes Treated with Metformin: A Randomised Glycaemia-Controlled Crossover Study. Diabetologia 2011, 54, 1799–1809.
- Jensterle, M.; Kravos, N.A.; Dolžan, V.; Goričar, K.; Herman, R.; Rizzo, M.; Janež, A. Glucose Transporter 4 MRNA Expression in Subcutaneous Adipose Tissue of Women with PCOS Remains Unchanged despite Metformin Withdrawal: Is There a Cellular Metabolic Treatment Legacy Effect? Endocrine 2021, 1–10. Available online: https://link.springer.com/article/10.1007%2Fs12020-021-02934-4 (accessed on 28 December 2021).
- Holmes, B.F.; Sparling, D.P.; Olson, A.L.; Winder, W.W.; Dohm, G.L. Regulation of Muscle GLUT4 Enhancer Factor and Myocyte Enhancer Factor 2 by AMP-Activated Protein Kinase. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E1071–E1076.
- Hardie, D.G. AMPK: A Target for Drugs and Natural Products with Effects on Both Diabetes and Cancer. Diabetes 2013, 62, 2164–2172.
- Lee, J.O.; Lee, S.K.; Jung, J.H.; Kim, J.H.; You, G.Y.; Kim, S.J.; Park, S.H.; Uhm, K.O.; Kim, H.S. Metformin Induces Rab4 through AMPK and Modulates GLUT4 Translocation in Skeletal Muscle Cells. J. Cell. Physiol. 2011, 226, 974–981.
- Lee, J.O.; Lee, S.K.; Kim, J.H.; Kim, N.; You, G.Y.; Moon, J.W.; Kim, S.J.; Park, S.H.; Kim, H.S. Metformin Regulates Glucose Transporter 4 (GLUT4) Translocation through AMP-Activated Protein Kinase (AMPK)-Mediated Cbl/CAP Signaling in 3T3-L1 Preadipocyte Cells. J. Biol. Chem. 2012, 287, 44121–44129.
- Singh, R.; Chandel, S.; Dey, D.; Ghosh, A.; Roy, S.; Ravichandiran, V.; Ghosh, D. Epigenetic Modification and Therapeutic Targets of Diabetes Mellitus. Biosci. Rep. 2020, 40, BSR20202160.
- Sookoian, S.; Pirola, C.J. Epigenetics of Insulin Resistance: An Emerging Field in Translational Medicine. Curr. Diab. Rep. 2013, 13, 229–237.
- Ling, C.; Groop, L. Epigenetics: A Molecular Link between Environmental Factors and Type 2 Diabetes. Diabetes 2009, 58, 2718–2725.
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic Effects of Metformin: From Molecular Mechanisms to Clinical Implications. Diabetes Obes. Metab. 2018, 20, 1553–1562.
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.-F.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin Regulates Global DNA Methylation via Mitochondrial One-Carbon Metabolism. Oncogene 2018, 37, 963–970.
- McGee, S.L.; van Denderen, B.J.W.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-Activated Protein Kinase Regulates GLUT4 Transcription by Phosphorylating Histone Deacetylase 5. Diabetes 2008, 57, 860–867.
- Yoshizaki, T.; Milne, J.C.; Imamura, T.; Schenk, S.; Sonoda, N.; Babendure, J.L.; Lu, J.-C.; Smith, J.J.; Jirousek, M.R.; Olefsky, J.M. SIRT1 Exerts Anti-Inflammatory Effects and Improves Insulin Sensitivity in Adipocytes. Mol. Cell. Biol. 2009, 29, 1363–1374.
- Arner, P.; Kulyté, A. MicroRNA Regulatory Networks in Human Adipose Tissue and Obesity. Nat. Rev. Endocrinol. 2015, 11, 276–288.
- Cirillo, F.; Catellani, C.; Lazzeroni, P.; Sartori, C.; Nicoli, A.; Amarri, S.; La Sala, G.B.; Street, M.E. MiRNAs Regulating Insulin Sensitivity Are Dysregulated in Polycystic Ovary Syndrome (PCOS) Ovaries and Are Associated With Markers of Inflammation and Insulin Sensitivity. Front. Endocrinol. (Lausanne) 2019, 10, 879.
- Esteves, J.V.; Enguita, F.J.; Machado, U.F. MicroRNAs-Mediated Regulation of Skeletal Muscle GLUT4 Expression and Translocation in Insulin Resistance. J. Diabetes Res. 2017, 2017, 7267910.
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297.
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-Mediated Increase in DICER1 Regulates MicroRNA Expression and Cellular Senescence. Aging Cell 2016, 15, 572–581.
- Hathaway, Q.; Pinti, M.; Durr, A.; Waris, S.; Shepherd, D.; Hollander, J. Regulating MicroRNA Expression: At the Heart of Diabetes Mellitus and the Mitochondrion. Am. J. Physiol.-Hear. Circ. Physiol. 2017, 314, ajpheart.00520.2017.
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin Elicits Anticancer Effects through the Sequential Modulation of DICER and C-MYC. Nat. Commun. 2012, 3, 865.
- Chen, Y.H.; Heneidi, S.; Lee, J.M.; Layman, L.C.; Stepp, D.W.; Gamboa, G.M.; Chen, B.S.; Chazenbalk, G.; Azziz, R. Mirna-93 Inhibits Glut4 and Is Overexpressed in Adipose Tissue of Polycystic Ovary Syndrome Patients and Women with Insulin Resistance. Diabetes 2013, 62, 2278–2286.
- Chuang, T.-Y.; Wu, H.-L.; Chen, C.-C.; Gamboa, G.M.; Layman, L.C.; Diamond, M.P.; Azziz, R.; Chen, Y.-H. MicroRNA-223 Expression Is Upregulated in Insulin Resistant Human Adipose Tissue. J. Diabetes Res. 2015, 2015, 943659.
- Zhou, T.; Meng, X.; Che, H.; Shen, N.; Xiao, D.; Song, X.; Liang, M.; Fu, X.; Ju, J.; Li, Y.; et al. Regulation of Insulin Resistance by Multiple MiRNAs via Targeting the GLUT4 Signalling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 38, 2063–2078.
- Esteves, J.V.; Yonamine, C.Y.; Pinto-Junior, D.C.; Gerlinger-Romero, F.; Enguita, F.J.; Machado, U.F. Diabetes Modulates MicroRNAs 29b-3p, 29c-3p, 199a-5p and 532-3p Expression in Muscle: Possible Role in GLUT4 and HK2 Repression. Front. Endocrinol. (Lausanne) 2018, 9, 536.
- Mysore, R.; Ortega, F.J.; Latorre, J.; Ahonen, M.; Savolainen-Peltonen, H.; Fischer-Posovszky, P.; Wabitsch, M.; Olkkonen, V.M.; Fernández-Real, J.M.; Haridas, P.A.N. MicroRNA-221-3p Regulates Angiopoietin-Like 8 (ANGPTL8) Expression in Adipocytes. J. Clin. Endocrinol. Metab. 2017, 102, 4001–4012.
- Massart, J.; Sjögren, R.J.O.; Lundell, L.S.; Mudry, J.M.; Franck, N.; O’Gorman, D.J.; Egan, B.; Zierath, J.R.; Krook, A. Altered MiR-29 Expression in Type 2 Diabetes Influences Glucose and Lipid Metabolism in Skeletal Muscle. Diabetes 2017, 66, 1807–1818.
- Wang, J.; Gao, Y.; Duan, L.; Wei, S.; Liu, J.; Tian, L.; Quan, J.; Zhang, Q.; Liu, J.; Yang, J. Metformin Ameliorates Skeletal Muscle Insulin Resistance by Inhibiting MiR-21 Expression in a High-Fat Dietary Rat Model. Oncotarget 2017, 8, 98029–98039.
- Mensà, E.; Giuliani, A.; Matacchione, G.; Gurau, F.; Bonfigli, A.R.; Romagnoli, F.; De Luca, M.; Sabbatinelli, J.; Olivieri, F. Circulating MiR-146a in Healthy Aging and Type 2 Diabetes: Age- and Gender-Specific Trajectories. Mech. Ageing Dev. 2019, 180.
- Roos, J.; Enlund, E.; Funcke, J.-B.; Tews, D.; Holzmann, K.; Debatin, K.-M.; Wabitsch, M.; Fischer-Posovszky, P. MiR-146a-Mediated Suppression of the Inflammatory Response in Human Adipocytes. Sci. Rep. 2016, 6, 38339.
- Balasubramanyam, M.; Aravind, S.; Gokulakrishnan, K.; Prabu, P.; Sathishkumar, C.; Ranjani, H.; Mohan, V. Impaired MiR-146a Expression Links Subclinical Inflammation and Insulin Resistance in Type 2 Diabetes. Mol. Cell. Biochem. 2011, 351, 197–205.
- Ortega, F.J.; Mercader, J.M.; Moreno-Navarrete, J.M.; Rovira, O.; Guerra, E.; Esteve, E.; Xifra, G.; Martínez, C.; Ricart, W.; Rieusset, J.; et al. Profiling of Circulating MicroRNAs Reveals Common MicroRNAs Linked to Type 2 Diabetes That Change with Insulin Sensitization. Diabetes Care 2014, 37, 1375–1383.
- Ghai, V.; Kim, T.-K.; Etheridge, A.; Nielsen, T.; Hansen, T.; Pedersen, O.; Galas, D.; Wang, K. Extracellular Vesicle Encapsulated MicroRNAs in Patients with Type 2 Diabetes Are Affected by Metformin Treatment. J. Clin. Med. 2019, 8, 617.
- Udesen, P.B.; Glintborg, D.; Sørensen, A.E.; Svendsen, R.; Nielsen, N.L.S.; Wissing, M.L.M.; Andersen, M.S.; Englund, A.L.M.; Dalgaard, L.T. Metformin Decreases MiR-122, MiR-223 and MiR-29a in Women with Polycystic Ovary Syndrome. Endocr. Connect. 2020, 9, 1075–1084.
- García-Calzón, S.; Perfilyev, A.; Martinell, M.; Ustinova, M.; Kalamajski, S.; Franks, P.W.; Bacos, K.; Elbere, I.; Pihlajamäki, J.; Volkov, P.; et al. Epigenetic Markers Associated with Metformin Response and Intolerance in Drug-Naïve Patients with Type 2 Diabetes. Sci. Transl. Med. 2020, 12, 561.
- Takahashi, N.; Kimura, A.P.; Otsuka, K.; Ohmura, K.; Naito, S.; Yoshida, M.; Ieko, M. Dreh, a Long Noncoding RNA Repressed by Metformin, Regulates Glucose Transport in C2C12 Skeletal Muscle Cells. Life Sci. 2019, 236, 116906.
- Li, P.; Tong, L.; Song, Y.; Sun, J.; Shi, J.; Wu, Z.; Diao, Y.; Li, Y.; Wang, Z. Long Noncoding RNA H19 Participates in Metformin-Mediated Inhibition of Gastric Cancer Cell Invasion. J. Cell. Physiol. 2019, 234, 4515–4527.
- Gui, W.; Zhu, W.F.; Zhu, Y.; Tang, S.; Zheng, F.; Yin, X.; Lin, X.; Li, H. LncRNAH19 Improves Insulin Resistance in Skeletal Muscle by Regulating Heterogeneous Nuclear Ribonucleoprotein A1. Cell Commun. Signal. 2020, 18, 173.
- Mueckler, M. Insulin Resistance and the Disruption of Glut4 Trafficking in Skeletal Muscle. J. Clin. Investig. 2001, 107, 1211–1213.
- Shepherd, P.R.; Kahn, B.B. Glucose Transporters and Insulin Action--Implications for Insulin Resistance and Diabetes Mellitus. N. Engl. J. Med. 1999, 341, 248–257.
- Xu, P.-T.; Song, Z.; Zhang, W.-C.; Jiao, B.; Yu, Z.-B. Impaired Translocation of GLUT4 Results in Insulin Resistance of Atrophic Soleus Muscle. Biomed Res. Int. 2015, 2015, 291987.
- Tremblay, F.; Lavigne, C.; Jacques, H.; Marette, A. Defective Insulin-Induced GLUT4 Translocation in Skeletal Muscle of High Fat-Fed Rats Is Associated with Alterations in Both Akt/Protein Kinase B and Atypical Protein Kinase C (Zeta/Lambda) Activities. Diabetes 2001, 50, 1901–1910.
- Goodyear, L.J.; Kahn, B.B. Exercise, Glucose Transport, and Insulin Sensitivity. Annu. Rev. Med. 1998, 49, 235–261.
- Shepherd, P.R.; Withers, D.J.; Siddle, K. Phosphoinositide 3-Kinase: The Key Switch Mechanism in Insulin Signalling. Biochem. J. 1998, 333, 471–490.
- Purintrapiban, J.; Ratanachaiyavong, S.; Mcshane, H. The Effects of Insulin and Metformin on Glucose Uptake in L8 Myotubes. ScienceAsia 2003, 29, 341.
- Watson, R.T.; Pessin, J.E. Bridging the GAP between Insulin Signaling and GLUT4 Translocation. Trends Biochem. Sci. 2006, 31, 215–222.
- Gao, L.; Chen, J.; Gao, J.; Wang, H.; Xiong, W. Super-Resolution Microscopy Reveals the Insulin-Resistance-Regulated Reorganization of GLUT4 on Plasma Membranes. J. Cell Sci. 2017, 130, 396–405.
- Corrêa-Giannella, M.L.; Machado, U.F. SLC2A4gene: A Promising Target for Pharmacogenomics of Insulin Resistance. Pharmacogenomics 2013, 14, 847–850.
- Fischer, Y.; Thomas, J.; Rösen, P.; Kammermeier, H. Action of Metformin on Glucose Transport and Glucose Transporter Glut1 and Glut4 in Heart Muscle Cells from Healthy and Diabetic Rats. Endocrinology 1995, 136, 412–420.
- Matthaei, S.; Hamann, A.; Klein, H.H.; Benecke, H.; Kreymann, G.; Flier, J.S.; Greten, H. Association of Metformin’s Effect to Increase Insulin-Stimulated Glucose Transport with Potentiation of Insulin-Induced Translocation of Glucose Transporters from Intracellular Pool to Plasma Membrane in Rat Adipocytes. Diabetes 1991, 40, 850–857.
- Sarabia, V.; Lam, L.; Burdett, E.; Leiter, L.A.; Klip, A. Glucose Transport in Human Skeletal Muscle Cells in Culture. Stimulation by Insulin and Metformin. J. Clin. Investig. 1992, 90, 1386–1395.
- Yang, J.; Holman, G.D. Long-Term Metformin Treatment Stimulates Cardiomyocyte Glucose Transport through an AMP-Activated Protein Kinase-Dependent Reduction in GLUT4 Endocytosis. Endocrinology 2006, 147, 2728–2736.
- Detaille, D.; Wiernsperger, N.; Devos, P. Metformin Interaction with Insulin-Regulated Glucose Uptake, Using the Xenopus Laevis Oocyte Model Expressing the Mammalian Transporter GLUT4. Eur. J. Pharmacol. 1999, 377, 127–136.
- Matthaei, S.; Reibold, J.P.; Hamann, A.; Benecke, H.; Häring, H.U.; Greten, H.; Klein, H.H. In Vivo Metformin Treatment Ameliorates Insulin Resistance: Evidence for Potentiation of Insulin-Induced Translocation and Increased Functional Activity of Glucose Transporters in Obese (Fa/Fa) Zucker Rat Adipocytes. Endocrinology 1993, 133, 304–311.
- Baus, D.; Heermeier, K.; De Hoop, M.; Metz-Weidmann, C.; Gassenhuber, J.; Dittrich, W.; Welte, S.; Tennagels, N. Identification of a Novel AS160 Splice Variant That Regulates GLUT4 Translocation and Glucose-Uptake in Rat Muscle Cells. Cell. Signal. 2008, 20, 2237–2246.
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-Stimulated Phosphorylation of a Rab GTPase-Activating Protein Regulates GLUT4 Translocation. J. Biol. Chem. 2003, 278, 14599–14602.
More