Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sankar Basu and Version 3 by Catherine Yang.
Structure-based drug design (SBDD) is the computational approach that relies on knowledge of 3D structures of the biological targets to identify or design the potential chemical structure suitable for clinical tests. With the explosion of genomic, functional, and structural information in recent decades, the majority of biological targets with 3D structure have been identified and stimulated the applications of structure-based approaches in the current design pipeline. SBDD is popular for virtual screening to filter the drug-like compounds from a large library of small molecules, including widely applied approaches, such as docking and structure-based pharmacophore design.
- mutations
- drug design
- macro-molecular docking
- pharmacophores
1. Docking
Docking is one common approach for compound screening. The basic idea is to use scoring functions [1][2][3] [1, 2, 3] to evaluate the fitness [4] [4] of the target protein in complex with the docked compound [5]. While sampling and scoring being the two major components of docking, a wide range of docking platforms are available to perform fast docking, offering a wide array of protocols (and scoring functions), such as Dock6 [6], Autodock Vina [7], Glide [8], Surflex [9], Cluspro [10] [10] and many others. The 3D atomic model (structural coordinates) of the target protein (receptor) is a pre-requisite to have for docking per se which could be either experimentally solved or computationally modeled. For the later case (in absence of an experimental template), one needs to do intensive modeling to generate the best representative structure or set of structures [11][12][11,12]. Such approaches are often adopted for IDPs / IDPRs in combined docking and dynamics in silico experiments [13][14][13, 14]. In the past, SBDD (Figure 1) has widely been used to mitigate the effects of non-synonymous (and/or deleterious) mutations related to many common diseases.
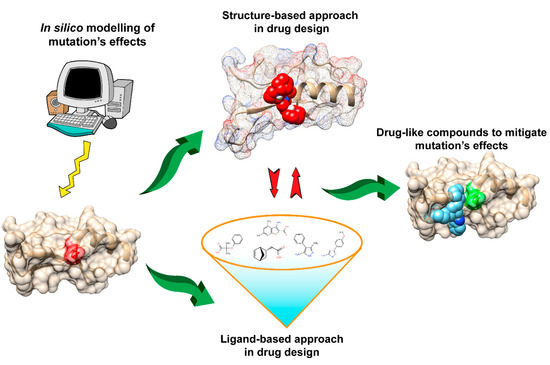 Figure 1. Schematic presentation of the drug discovery process to mitigate the effects of disease-causing mutations.
Examples include p53, an enigmatic multi-functional protein with intrinsically disordered regions [15], the so-called “guardian protein” in cancer, functioning as a tumor-suppressor [16]. Again, there are specific mutations in p53 that result in the malfunctioning of the protein and increase the risk of cancers [17]. In cancer patients, mutations acting against p53 in its DNA binding ability (in its role of a transcription factor) are frequently observed. Rescuing (or supplementing) the native function(s) in the ‘mutant p53 protein’ is one central objective in current cancer research [18][19][18,19]. In the past, it has been shown that the small molecules can be designed to bind to an adaptive protein–protein interface [20] [20] and can be made to stabilize the DNA binding domain and rescue mutant functions [9][21]. Recent computational recent in an aligned direction modeled the wild-type p53 and several of its deleterious mutants [21] [22] shaded light on the mechanism of p53 reactivation [21][22]. A novel transiently open L1/L3 pocket was identified and indicated the exposure of Cys-124 was pivotal in the formation of cavities [21][22]. It was a crucial finding since Cys-124 was suggested to be the 'covalent' docking site for known alkylating p53 stabilizers [22][23]. This made interest and scope to pursue a thorough screening of potential compounds, docked onto this pocket to search for other potential stabilizers. As a matter of fact, 1,324 compounds from the NCI/DTP Open Chemical Repository Diversity Set II were docked onto the generated ensemble structures of the R273H cancer mutant, out of which 45 compounds were chosen for biological assay [21][22]. Finally, stictic acid (NSC-87511) (Figure 2) was experimentally validated to be an efficient reactivation compound for mutant p53 [21][22].
Figure 1. Schematic presentation of the drug discovery process to mitigate the effects of disease-causing mutations.
Examples include p53, an enigmatic multi-functional protein with intrinsically disordered regions [15], the so-called “guardian protein” in cancer, functioning as a tumor-suppressor [16]. Again, there are specific mutations in p53 that result in the malfunctioning of the protein and increase the risk of cancers [17]. In cancer patients, mutations acting against p53 in its DNA binding ability (in its role of a transcription factor) are frequently observed. Rescuing (or supplementing) the native function(s) in the ‘mutant p53 protein’ is one central objective in current cancer research [18][19][18,19]. In the past, it has been shown that the small molecules can be designed to bind to an adaptive protein–protein interface [20] [20] and can be made to stabilize the DNA binding domain and rescue mutant functions [9][21]. Recent computational recent in an aligned direction modeled the wild-type p53 and several of its deleterious mutants [21] [22] shaded light on the mechanism of p53 reactivation [21][22]. A novel transiently open L1/L3 pocket was identified and indicated the exposure of Cys-124 was pivotal in the formation of cavities [21][22]. It was a crucial finding since Cys-124 was suggested to be the 'covalent' docking site for known alkylating p53 stabilizers [22][23]. This made interest and scope to pursue a thorough screening of potential compounds, docked onto this pocket to search for other potential stabilizers. As a matter of fact, 1,324 compounds from the NCI/DTP Open Chemical Repository Diversity Set II were docked onto the generated ensemble structures of the R273H cancer mutant, out of which 45 compounds were chosen for biological assay [21][22]. Finally, stictic acid (NSC-87511) (Figure 2) was experimentally validated to be an efficient reactivation compound for mutant p53 [21][22].
Figure 1. Schematic presentation of the drug discovery process to mitigate the effects of disease-causing mutations.
Examples include p53, an enigmatic multi-functional protein with intrinsically disordered regions [15], the so-called “guardian protein” in cancer, functioning as a tumor-suppressor [16]. Again, there are specific mutations in p53 that result in the malfunctioning of the protein and increase the risk of cancers [17]. In cancer patients, mutations acting against p53 in its DNA binding ability (in its role of a transcription factor) are frequently observed. Rescuing (or supplementing) the native function(s) in the ‘mutant p53 protein’ is one central objective in current cancer research [18][19][18,19]. In the past, it has been shown that the small molecules can be designed to bind to an adaptive protein–protein interface [20] [20] and can be made to stabilize the DNA binding domain and rescue mutant functions [9][21]. Recent computational recent in an aligned direction modeled the wild-type p53 and several of its deleterious mutants [21] [22] shaded light on the mechanism of p53 reactivation [21][22]. A novel transiently open L1/L3 pocket was identified and indicated the exposure of Cys-124 was pivotal in the formation of cavities [21][22]. It was a crucial finding since Cys-124 was suggested to be the 'covalent' docking site for known alkylating p53 stabilizers [22][23]. This made interest and scope to pursue a thorough screening of potential compounds, docked onto this pocket to search for other potential stabilizers. As a matter of fact, 1,324 compounds from the NCI/DTP Open Chemical Repository Diversity Set II were docked onto the generated ensemble structures of the R273H cancer mutant, out of which 45 compounds were chosen for biological assay [21][22]. Finally, stictic acid (NSC-87511) (Figure 2) was experimentally validated to be an efficient reactivation compound for mutant p53 [21][22].
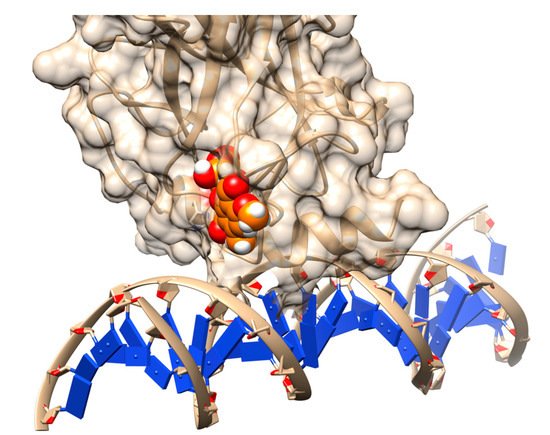
Figure 2. Rescuing the activity of mutant p53 by binding stictic acid into the open L1/L3 pocket. The representative scheme is generated using the structure of p53 core domain complex with DNA (PDB: 1TSR). Atoms in sphere representation belong to stictic acid in a given docked pose. DNA bases are represented as blue squares.
Besides cancer research, docking based screening has also been used in other rare diseases. For example, Snyder–Robinson Syndrome (SRS) is a rare X-chromosome linked mental disease, caused by a malfunctioning spermine synthase [23][24]. Spermine synthase functions as homo-dimer and mutations affecting the dimerization such as G56S are shown to abolish the enzyme activity to result in the disease [23][24][24, 25]. Recent studies targeted identification of dimer stabilizers by binding to the mutant homo-dimer interface [25][26]. Integrated large commercial compound libraries were used for this docking-based virtual screening with the representative structures of the dimer [25][26]. The best-ranked 51 compounds were then subjected to experimental screening out of which three top-ranked compounds (also known as ‘leads’) was shown to enhance the catalytic activity of Spermine synthase up to 30% [25][26][26, 27].
2. Structure-Based Pharmacophore Design
Pharmacophore models can be used to make an ensemble of abstract steric and electronic features representing macromolecular (target protein) interactions with drug-like small molecules [27][28][28, 29]. In other words, three-dimensional arrangements of these features such as hydrophobic centroids, aromatic rings and hydrogen bonds are representation of the binding mode between the ligand and the target [28][29][29, 30]. Pharmacophores are generated from common features of active ligands, which are identified by aligning or superimposing the conformers of either ligand-target complexes or known active molecules [29][30]. Multiple degenerate atomic models can potentially be output from pharmacophore modeling programs requiring further optimization and validation to select the best one. Pharmacophore models are commonly used for virtual screening of active small molecules from large compound databases [27][28][29][28, 29, 30]. Such approaches can be more efficient than docking for certain targets, especially when a large number of existing known active compounds are available [30][31][32][31, 32, 33].
Pharmacophore models have also been used to identify active molecules to mitigate the effects of mutations in many diseases [34, [33][34]35]. For cases where a sufficient number of active molecules are previously known for generating high-quality pharmacophore models, pharmacophore proves to be a powerful tool for drug ‘lead’ identification [35][36]. Recent work has applied structure-based pharmacophore analysis to identify the novel ROS-1 inhibitors to curb the drug resistance problem caused by mutations [33][37]. Proto-oncogene receptor tyrosine kinase ROS-1 is ectopicly and oncogenicly expressed in many cancers, mainly in non-small cell lung cancer (NSCLC) [33][37]. ROS-1 is highly homologous with the kinase domain of anaplastic lymphoma kinase (ALK) and FDA approved ALK inhibitors such as Crizotinib are experimentally validated as therapeutics against ROS-1 driven tumors [33][37]. However, these commercial ROS-1 inhibitors lack a broad spectrum of activity due to the growing resistance from ROS-1 mutations, primarily G2032R [34][38]. Following on, a pharmacophore model was built using the complex structure of both wildtype (WT) and mutant ROS-1 with previously known inhibitors to identify more general inhibitors against both WT and mutant [36][37][39, 40]. Pharmacophore-based virtual screening was then performed to selected candidates from commercial databases with further filtering and scoring analysis. Five hits were eventually identified with good binding affinities to both WT and mutant [37][40].
Thus, pharmacophore essentially defines the interaction framework among the active ligands, and their specific targets [27][28] [28, 29] and the corresponding models can also be built with libraries of active ligands alone, in the absence of the 3D structure of the target—an approach known as ligand based pharmacophore. The models, therein, can then be trained for discrimination between active and inactive molecules [21][28]. In fact, this serves as the prime reason of widespread use of pharmacophore models in virtual screening especially when lacking the target structure. In addition, as the pharmacophore model represents the binding (or interaction map) of ‘active compounds-target interaction’, it provides a plausible relationship between the structure and the ligand activity and could help to elucidate the underlying biochemical mechanism to further guide the design of the novel active compounds [22][29]. For example, by exploring the different pharmacological properties, recent studies have seemed to improve the potency of existing pharmacophore and designed novel epidermal growth factor receptor (EGFR) inhibitor potentially inhibited by primary mutants (L858R, del9) and drug-resistant mutants, such as L858R/T790M [34][38].