The tumor necrosis factor (TNF) ligand family includes 19 ligands, which each contain a C-terminal TNF homology domain (THD). Each ligand binds to one or multiple TNF receptors (TNFR), containing extracellular cysteine-rich domains (CRD) for ligand binding. Tumor necrosis factor (TNF or TNF-α) and lymphotoxin (LT) are the first two cytokines that have been characterized as TNF superfamily members. They have a homologous amino acid sequence and were both discovered based on their anti-tumor effects. Moreover, both ligands bind to TNF receptor type 1 and type 2 (TNFR1 and TNFR2) and mediate similar cell signaling transduction.
- TNF family
- protein engineering
- receptor specificity
- ligand
- TNF-α
1. Introduction
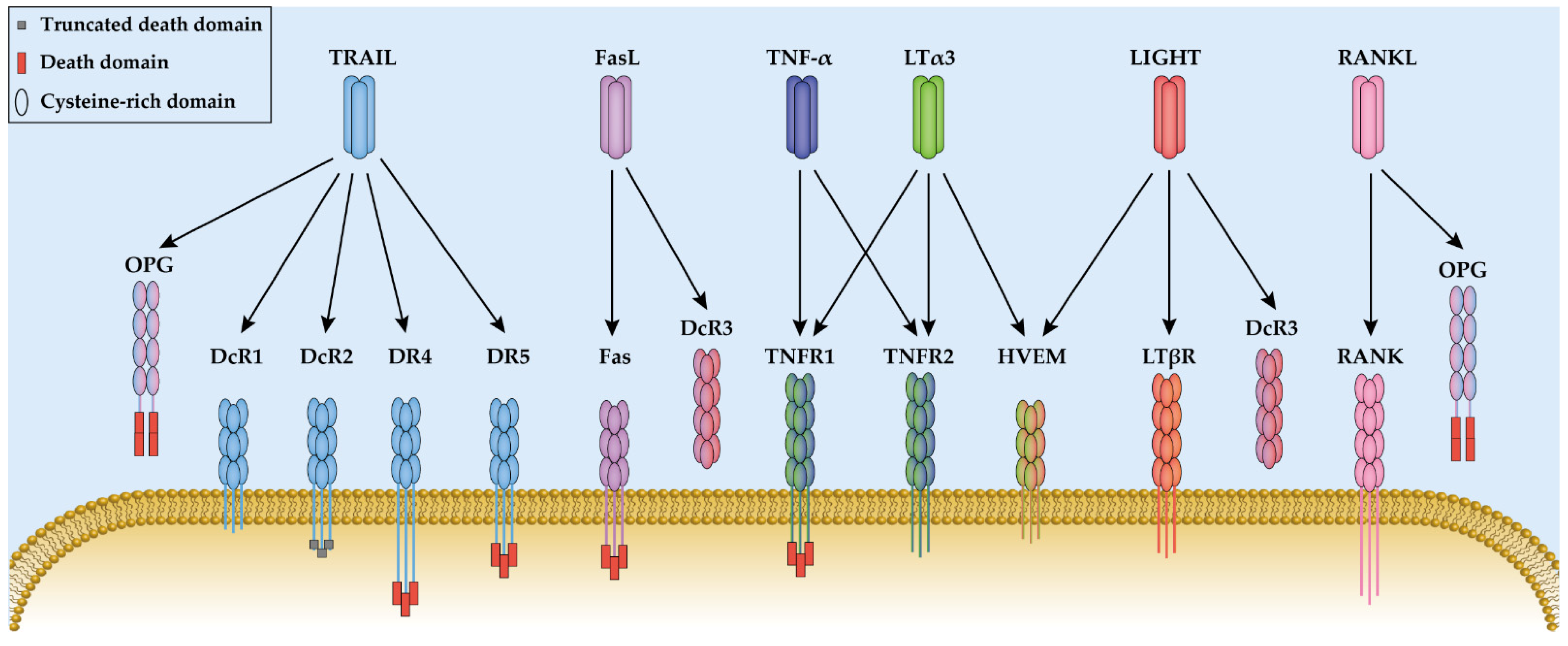
2. Tumor Necrosis Factor and Lymphotoxin
2.1. Tumor Necrosis Factor and Lymphotoxin
2.2. The TNFR1 and TNFR2 Induced Signal Transduction
2.3. Engineering TNFR1-Specific Ligands
Ligand | Specificity | Variants | Mutation Sites | Binding Affinity (nM) | Ref. | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
TNFR1 | TNFR2 | |||||||||||||||||||
TNF-α | TNFR1 &TNFR2 | WT | / | 15.8 | 35.3 |
[41] |
||||||||||||||
TNFR1 | L29S | L29S | − 1 | − |
[42] |
|||||||||||||||
R32W | R32W | − | − |
[42] |
||||||||||||||||
R32W-S86T | R32W/S86T | 3540 | NB 2 |
[42] |
||||||||||||||||
F4614 | T5G/P6D/R29V | − | − |
[38] |
||||||||||||||||
M3S | L29S/S52I/Y56F | and 449/455del | − | − |
[40] |
|||||||||||||||
mutTNF G4 | A84S/V85S/S86T/ | Q88N/T89P | 8.72 | NB |
[41] |
|||||||||||||||
TNFR2 | D143N-A145R | D143N/A145R | NB 2 | 13.1 |
[42] |
|||||||||||||||
2.4. TNFR2-Specific Applications
References
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756.
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160.
- Croft, M.; Siegel, R.M. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 217–233.
- Anti-TNFα Use During Elective Foot and Ankle Surgery in Patients With Rheumatoid Arthritis. Available online: https://clinicaltrials.gov/ct2/show/NCT02242474?term=TNF-α&draw=3&rank=11 (accessed on 5 January 2022).
- Targeted Stem Cells Expressing TRAIL as a Therapy for Lung Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03298763?term=TRAIL&draw=2&rank=7 (accessed on 5 January 2022).
- Safety, Tolerability and PK/PD of JMT103 in Patients With Bone Metastases From Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03550508?term=RANKL&draw=2&rank=13 (accessed on 5 January 2022).
- Determination of the RANKL/Osteoprotegerin Ratio in Patients With Systemic Lupus Erythematosus. Role in Osteoporosis and Cardiovascular Calcification. Available online: https://clinicaltrials.gov/ct2/show/NCT02799173?term=RANKL&draw=2&rank=4 (accessed on 5 January 2022).
- Buhrmann, C.; Shayan, P.; Aggarwal, B.B.; Shakibaei, M. Evidence that TNF-β (lymphotoxin α) can activate the inflammatory environment in human chondrocytes. Arthritis Res. Ther. 2013, 15, R202.
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666.
- Wang, J.; Wakeham, J.; Harkness, R.; Xing, Z. Macrophages are a significant source of type 1 cytokines during mycobacterial infection. J. Clin. Investig. 1999, 103, 1023–1029.
- Maskos, K.; Fernandez-Catalan, C.; Huber, R.; Bourenkov, G.P.; Bartunik, H.; Ellestad, G.A.; Reddy, P.; Wolfson, M.F.; Rauch, C.T.; Castner, B.J.; et al. Crystal structure of the catalytic domain of human tumor necrosis factor-α-converting enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 3408–3412.
- Dittrich, G.M.; Heineke, J. TNF-α signaling: TACE inhibition to put out the burning heart. PLoS Biol. 2020, 18, e3001037.
- Grell, M.; Wajant, H.; Zimmermann, G.; Scheurich, P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1998, 95, 570–575.
- Ruddle, N.H.; Waksman, B.H. Cytotoxicity mediated by soluble antigen and lymphocytes in delayed hypersensitivity. 3. Analysis of mechanism. J. Exp. Med. 1968, 128, 1267–1279.
- Kolb, W.P.; Granger, G.A. Lymphocyte in vitro cytotoxicity: Characterization of human lymphotoxin. Proc. Natl. Acad. Sci. USA 1968, 61, 1250–1255.
- Shalaby, M.R.; Aggarwal, B.B.; Rinderknecht, E.; Svedersky, L.P.; Finkle, B.S.; Palladino, M.A. Activation of human polymorphonuclear neutrophil functions by interferon-gamma and tumor necrosis factors. J. Immunol. 1985, 135, 2069–2073.
- Kucka, K.; Lang, I.; Zhang, T.; Siegmund, D.; Medler, J.; Wajant, H. Membrane lymphotoxin-α2β is a novel tumor necrosis factor (TNF) receptor 2 (TNFR2) agonist. Cell Death Dis. 2021, 12, 360.
- Gubernatorova, E.O.; Polinova, A.I.; Petropavlovskiy, M.M.; Namakanova, O.A.; Medvedovskaya, A.D.; Zvartsev, R.V.; Telegin, G.B.; Drutskaya, M.S.; Nedospasov, S.A. Dual role of tnf and ltα in carcinogenesis as implicated by studies in mice. Cancers 2021, 13, 1775.
- Roach, D.R.; Briscoe, H.; Saunders, B.; France, M.P.; Riminton, S.; Britton, W.J. Secreted lymphotoxin-alpha is essential for the control of an intracellular bacterial infection. J. Exp. Med. 2001, 193, 239–246.
- Kruglov, A.A.; Grivennikov, S.I.; Kuprash, D.V.; Winsauer, C.; Prepens, S.; Seleznik, G.M.; Eberl, G.; Littman, D.R.; Heikenwalder, M.; Tumanov, A.V.; et al. Nonredundant function of soluble ltα3 produced by innate lymphoid cells in intestinal homeostasis. Science 2013, 342, 1243–1246.
- Etemadi, N.; Holien, J.K.; Chau, D.; Dewson, G.; Murphy, J.M.; Alexander, W.S.; Parker, M.W.; Silke, J.; Nachbur, U. Lymphotoxin α induces apoptosis, necroptosis and inflammatory signals with the same potency as tumour necrosis factor. FEBS J. 2013, 280, 5283–5297.
- Pegoretti, V.; Baron, W.; Laman, J.D.; Eisel, U.L.M. Selective Modulation of TNF–TNFRs Signaling: Insights for Multiple Sclerosis Treatment. Front. Immunol. 2018, 9, 925.
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91.
- Raeiszadeh, M.; Verney, M.; Craddock, C.; Wajant, H.; Moss, P.; Chen, F. TNFR2 Is Expressed Preferentially By Late Differentiated CD8 T-Cells and Can be Triggered By TNFR2-Specific Ligand to Induce Cell Death of Recently Activated Antigen-Specific T Cells: A Possible Role of TNFR2 in T-Cell Deflation. Blood 2014, 124, 4352.
- Hijdra, D.; Vorselaars, A.D.; Grutters, J.C.; Claessen, A.M.; Rijkers, G.T. Differential expression of TNFR1 (CD120a) and TNFR2 (CD120b) on subpopulations of human monocytes. J. Inflamm. 2012, 9, 38.
- Beldi, G.; Bahiraii, S.; Lezin, C.; Nouri Barkestani, M.; Abdelgawad, M.E.; Uzan, G.; Naserian, S. TNFR2 Is a Crucial Hub Controlling Mesenchymal Stem Cell Biological and Functional Properties. Front. Cell Dev. Biol. 2020, 8, 596831.
- Naserian, S.; Shamdani, S.; Arouche, N.; Uzan, G. Regulatory T cell induction by mesenchymal stem cells depends on the expression of TNFR2 by T cells. Stem Cell Res. Ther. 2020, 11, 534.
- Morton, P.E.; Perrin, C.; Levitt, J.; Matthews, D.R.; Marsh, R.J.; Pike, R.; McMillan, D.; Maloney, A.; Poland, S.; Ameer-Beg, S.; et al. TNFR1 membrane reorganization promotes distinct modes of TNFα signaling. Sci. Signal. 2019, 12, 2418.
- Cabal-Hierro, L.; Lazo, P.S. Signal transduction by tumor necrosis factor receptors. Cell. Signal. 2012, 24, 1297–1305.
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190.
- Xu, D.; Zhao, H.; Jin, M.; Zhu, H.; Shan, B.; Geng, J.; Dziedzic, S.A.; Amin, P.; Mifflin, L.; Naito, M.G.; et al. Modulating TRADD to restore cellular homeostasis and inhibit apoptosis. Nature 2020, 587, 133–138.
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401.
- Van Ostade, X.; Vandenabeele, P.; Everaerdt, B.; Loetscher, H.; Gentz, R.; Brockhaus, M.; Lesslauer, W.; Tavernier, J.; Brouckaert, P.; Fiers, W. Human TNF mutants with selective activity on the p55 receptor. Nature 1993, 361, 266–269.
- Barbara, J.A.; Smith, W.B.; Gamble, J.R.; Van Ostade, X.; Vandenabeele, P.; Tavernier, J.; Fiers, W.; Vadas, M.A.; Lopez, A.F. Dissociation of TNF-alpha cytotoxic and proinflammatory activities by p55 receptor- and p75 receptor-selective TNF-alpha mutants. EMBO J. 1994, 13, 843–850.
- Loetscher, H.; Stueber, D.; Banner, D.; Mackay, F.; Lesslauer, W. Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J. Biol. Chem. 1993, 268, 26350–26357.
- Hube, F.; Hauner, H. The two tumor necrosis factor receptors mediate opposite effects on differentiation and glucose metabolism in human adipocytes in primary culture. Endocrinology 2000, 141, 2582–2588.
- Lees, D.M.; Pallikaros, Z.; Corder, R. The p55 tumor necrosis factor receptor (CD120a) induces endothelin-1 synthesis in endothelial and epithelial cells. Eur. J. Pharmacol. 2000, 390, 89–94.
- Shikama, H.; Miyata, K.; Sakae, N.; Mitsuishi, Y.; Nishimura, K.; Kuroda, K.; Kato, M. Novel mutein of tumor necrosis factor alpha (F4614) with reduced hypotensive effect. J. Interferon Cytokine Res. 1995, 15, 677–684.
- Atarashi, Y.; Yasumura, S.; Nambu, S.; Yoshio, Y.; Murakami, J.; Takahara, T.; Higuchi, K.; Watanabe, A.; Miyata, K.; Kato, M. A novel human tumor necrosis factor alfa mutein, F4614, inhibits in vitro and in vivo growth of murine and human hepatoma: Implication for immunotherapy of human hepatocellular carcinoma. Hepatology 1998, 28, 57–67.
- Cha, S.-S.; Kim, J.-S.; Cho, H.-S.; Shin, N.-K.; Jeong, W.; Shin, H.-C.; Kim, Y.J.; Hahn, J.H.; Oh, B.-H. High Resolution Crystal Structure of a Human Tumor Necrosis Factor-α Mutant with Low Systemic Toxicity. J. Biol. Chem. 1998, 273, 2153–2160.
- Munoz Pinto, M.F.; Campbell, S.J.; Simoglou Karali, C.; Johanssen, V.A.; Bristow, C.; Cheng, V.W.T.; Zarghami, N.; Larkin, J.R.; Pannell, M.; Hearn, A.; et al. Selective blood-brain barrier permeabilization of brain metastases by a type 1 receptor-selective tumor necrosis factor mutein. Neuro-Oncology 2021, 24, 52–63.
- Van Ostade, X.; Tavernier, J.; Prangé, T.; Fiers, W. Localization of the active site of human tumour necrosis factor (hTNF) by mutational analysis. EMBO J. 1991, 10, 827–836.
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527.
- Lee, J.U.; Shin, W.; Son, J.Y.; Yoo, K.-Y.; Heo, Y.-S. Molecular Basis for the Neutralization of Tumor Necrosis Factor α by Certolizumab Pegol in the Treatment of Inflammatory Autoimmune Diseases. Int. J. Mol. Sci. 2017, 18, 228.
- Genovese, M.C.; Cohen, S.; Moreland, L.; Lium, D.; Robbins, S.; Newmark, R.; Bekker, P. Combination Therapy with Etanercept and Anakinra in the Treatment of Patients with Rheumatoid Arthritis Who Have Been Treated Unsuccessfully with Methotrexate. Arthritis Rheum. 2004, 50, 1412–1419.
- Goodall, L.J.; Ovecka, M.; Rycroft, D.; Friel, S.L.; Sanderson, A.; Mistry, P.; Davies, M.L.; Stoop, A.A. Pharmacokinetic and pharmacodynamic characterisation of an anti-mouse TNF receptor 1 domain antibody formatted for in vivo half-life extension. PLoS ONE 2015, 10, e0137065.
- Gouweleeuw, L.; Wajant, H.; Maier, O.; Eisel, U.L.M.; Blankesteijn, W.M.; Schoemaker, R.G. Effects of selective TNFR1 inhibition or TNFR2 stimulation, compared to non-selective TNF inhibition, on (neuro)inflammation and behavior after myocardial infarction in male mice. Brain Behav. Immun. 2021, 93, 156–171.
- McCann, F.E.; Perocheau, D.P.; Ruspi, G.; Blazek, K.; Davies, M.L.; Feldmann, M.; Dean, J.L.E.; Stoop, A.A.; Williams, R.O. Selective tumor necrosis factor receptor i blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 2014, 66, 2728–2738.
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; Pick, L.; Haar, D.; Musiol, S.; Storch, M.K.; et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS ONE 2014, 9, 90117.
- Zettlitz, K.A.; Lorenz, V.; Landauer, K.; Münkel, S.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. MAbs 2010, 2, 639–647.
- Richter, F.; Liebig, T.; Guenzi, E.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. Antagonistic TNF Receptor One-Specific Antibody (ATROSAB): Receptor Binding and In Vitro Bioactivity. PLoS ONE 2013, 8, e72156.
- Chen, S.; Feng, Z.; Wang, Y.; Ma, S.; Hu, Z.; Yang, P.; Chai, Y.; Xie, X. Discovery of Novel Ligands for TNF-α and TNF Receptor-1 through Structure-Based Virtual Screening and Biological Assay. J. Chem. Inf. Model. 2017, 57, 1101–1111.
- Saddala, M.S.; Huang, H. Identification of novel inhibitors for TNFα, TNFR1 and TNFα-TNFR1 complex using pharmacophore-based approaches. J. Transl. Med. 2019, 17, 215.
- Lo, C.H.; Schaaf, T.M.; Grant, B.D.; Lim, C.K.W.; Bawaskar, P.; Aldrich, C.C.; Thomas, D.D.; Sachs, J.N. Noncompetitive inhibitors of TNFR1 probe conformational activation states. Sci. Signal. 2019, 12, 5637.
- Van De Kar, N.C.A.J.; Kooistra, T.; Vermeer, M.; Lesslauer, W.; Monnens, L.A.H.; Van Hinsbergh, V.W.M. Tumor necrosis factor α induces endothelial galactosyl transferase activity and verocytotoxin receptors. Role of specific tumor necrosis factor receptors and protein kinase C. Blood 1995, 85, 734–743.
- Yui, J.; Hemmings, D.; Garcia-Lloret, M.; Guilbert, L.J. Expression of the human p55 and p75 tumor necrosis factor receptors in primary villous trophoblasts and their role in cytotoxic signal transduction. Biol. Reprod. 1996, 55, 400–409.
- Xiao, J.; Garcia-Lloret, M.; Winkler-Lowen, B.; Miller, R.; Simpson, K.; Guilbert, L.J. ICAM-1-mediated adhesion of peripheral blood monocytes to the maternal surface of placental syncytiotrophoblasts: Implications for placental villitis. Am. J. Pathol. 1997, 150, 1845–1860.
- Dong, Y.; Fischer, R.; Naudé, P.J.W.; Maier, O.; Nyakas, C.; Duffey, M.; Van Der Zee, E.A.; Dekens, D.; Douwenga, W.; Herrmann, A.; et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 12304–12309.
- Fischer, R.; Proske, M.; Duffey, M.; Stangl, H.; Martinez, G.F.; Peters, N.; Kraske, A.; Straub, R.H.; Bethea, J.R.; Kontermann, R.E.; et al. Selective Activation of Tumor Necrosis Factor Receptor II Induces Antiinflammatory Responses and Alleviates Experimental Arthritis. Arthritis Rheumatol. 2018, 70, 722–735.
- Chopra, M.; Biehl, M.; Steinfatt, T.; Brandl, A.; Kums, J.; Amich, J.; Vaeth, M.; Kuen, J.; Holtappels, R.; Podlech, J.; et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med. 2016, 213, 1881–1900.
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS ONE 2011, 6, e27621.