Diabetes mellitus (DM) has been associated with cognitive complications in the brain resulting from acute and chronic metabolic disturbances happening peripherally and centrally. Numerous studies have reported on the morphological, electrophysiological, biochemical, and cognitive changes in the brains of diabetic individuals. The detailed pathophysiological mechanisms implicated in the development of the diabetic cognitive phenotype remain unclear due to intricate molecular changes evolving over time and space. This study provides an insight into recent advances in understanding molecular events in the diabetic brain, focusing on cerebral glucose and insulin uptake, insulin action in the brain, and the role of the brain in the regulation of glucose homeostasis. Fully competent mitochondria are essential for energy metabolism and proper brain function; hence, the potential contribution of mitochondria to the DM-induced impairment of the brain is also discussed.
- diabetes
- brain
- mitochondria
- proteostasis
1. Introduction
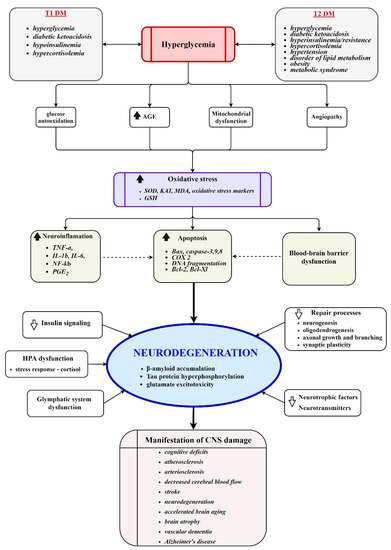
2. Insulin–Mitochondria–ROS Interplay
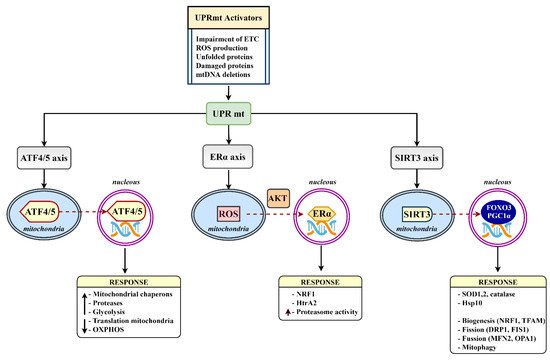
3. Mitochondrial Proteostasis in the Diabetic Brain
References
- Sima, A.A.F. Encephalopathies: The emerging diabetic complications. Acta Diabetol. 2010, 47, 279–293.
- Nouwen, A.; Chambers, A.; Chechlacz, M.; Higgs, S.; Blissett, J.; Barrett, T.; Allen, H.A. Microstructural abnormalities in white and gray matter in obese adolescents with and without type 2 diabetes. NeuroImage Clin. 2017, 16, 43–51.
- Chen, Z.; Li, L.; Sun, J.; Ma, L. Mapping the brain in type II diabetes: Voxel-based morphometry using DARTEL. Eur. J. Radiol. 2012, 81, 1870–1876.
- Mangia, S.; Kumar, A.F.; Moheet, A.; Roberts, R.J.; Eberly, L.; Seaquist, E.R.; Tkac, I. Neurochemical Profile of Patients with Type 1 Diabetes Measured by 1H-MRS at 4 T. Br. J. Pharmacol. 2013, 33, 754–759.
- Lin, A.; Northam, E.A.; Rankins, D.; Werther, G.A.; Cameron, F.J. Neuropsychological profiles of young people with type 1 diabetes 12 yr after disease onset. Pediatr. Diabetes 2010, 11, 235–243.
- van Duinkerken, E.; Ryan, C.M. Diabetes mellitus in the young and the old: Effects on cognitive functioning across the life span. Neurobiol. Dis. 2020, 134, 104608.
- Marseglia, A.; Fratiglioni, L.; Laukka, E.J.; Santoni, G.; Pedersen, N.L.; Bäckman, L.; Xu, W. Early Cognitive Deficits in Type 2 Diabetes: A Population-Based Study. J. Alzheimer’s Dis. 2016, 53, 1069–1078.
- Chatterjee, S.; Peters, S.; Woodward, M.; Arango, S.M.; Batty, G.; Beckett, N.; Beiser, A.; Borenstein, A.R.; Crane, P.K.; Haan, M.N.; et al. Type 2 Diabetes as a Risk Factor for Dementia in Women Compared with Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes Care 2015, 39, 300–307.
- Smolina, K.; Wotton, C.; Goldacre, M.J. Risk of dementia in patients hospitalized with type 1 and type 2 diabetes in England, 1998–2011: A retrospective national record linkage cohort study. Diabetologia 2015, 58, 942–950.
- You, Y.; Liu, Z.; Chen, Y.; Xu, Y.; Qin, J.; Guo, S.; Huang, J.; Tao, J. The prevalence of mild cognitive impairment in type 2 diabetes mellitus patients: A systematic review and meta-analysis. Acta Diabetol. 2021, 58, 671–685.
- Assunção, N.; Sudo, F.K.; Drummond, C.; De Felice, F.G.; Mattos, P. Metabolic Syndrome and cognitive decline in the elderly: A systematic review. PLoS ONE 2018, 13, e0194990.
- Ruegsegger, G.N.; Manjunatha, S.; Summer, P.; Gopala, S.; Zabeilski, P.; Dasari, S.; Vanderboom, P.M.; Lanza, I.R.; Klaus, K.A.; Nair, K.S. Insulin deficiency and intranasal insulin alter brain mitochondrial function: A potential factor for dementia in diabetes. FASEB J. 2019, 33, 4458–4472.
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2010, 53, 160–169.
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 9, 496.
- Persiyantseva, N.A.; Storozhevykh, T.P.; Senilova, Y.E.; Gorbacheva, L.R.; Pinelis, V.G.; Pomytkin, I.A. Mitochondrial H2O2 as an enable signal for triggering autophosphorylation of insulin receptor in neurons. J. Mol. Signal. 2013, 8, 11.
- Bolaños, J.P.; Almeida, A.; Moncada, S. Glycolysis: A bioenergetic or a survival pathway? Trends Biochem. Sci. 2010, 35, 145–149.
- Chomova, M.; Zitnanova, I. Look into brain energy crisis and membrane pathophysiology in ischemia and reperfusion. Stress 2016, 19, 341–348.
- Kamboj, S.S.; Sandhir, R. Protective effect of N-acetylcysteine supplementation on mitochondrial oxidative stress and mitochondrial enzymes in cerebral cortex of streptozotocin-treated diabetic rats. Mitochondrion 2011, 11, 214–222.
- Chomova, M.; Balazova, M.; Muchova, J. Diabetes-induced abnormalities of mitochondrial function in rat brain cortex: The effect of n-3 fatty acid diet. Mol. Cell. Biochem. 2017, 435, 109–131.
- Poulsen, H.E.; Weimann, A.; Henriksen, T.; Kjær, L.K.; Larsen, E.L.; Carlsson, E.R.; Christensen, C.K.; Brandslund, I.; Fenger, M. Oxidatively generated modifications to nucleic acids in vivo: Measurement in urine and plasma. Free Radic. Biol. Med. 2019, 145, 336–341.
- Carvalho, C.; Santos, M.S.; Oliveira, C.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta. 2015, 1852, 1665–1675.
- Chowdhury, S.R.; Saleh, A.; Akude, E.; Smith, D.R.; Morrow, D.; Tessler, L.; Calcutt, N.A.; Fernyhough, P. Ciliary Neurotrophic Factor Reverses Aberrant Mitochondrial Bioenergetics Through the JAK/STAT Pathway in Cultured Sensory Neurons Derived from Streptozotocin-Induced Diabetic Rodents. Cell. Mol. Neurobiol. 2014, 34, 643–649.
- Lu, H.; Koshkin, V.; Allister, E.M.; Gyulkhandanyan, A.V.; Wheeler, M.B. Molecular and Metabolic Evidence for Mitochondrial Defects Associated With β-Cell Dysfunction in a Mouse Model of Type 2 Diabetes. Diabetes 2009, 59, 448–459.
- Gerencser, A.A. Bioenergetic Analysis of Single Pancreatic β-Cells Indicates an Impaired Metabolic Signature in Type 2 Diabetic Subjects. Endocrinology 2015, 156, 3496–3503.
- Siewiera, K.; Kassassir, H.; Talar, M.; Wieteska, L.; Watala, C. Higher mitochondrial potential and elevated mitochondrial respiration are associated with excessive activation of blood platelets in diabetic rats. Life Sci. 2016, 148, 293–304.
- Cheng, G.; Zielonka, M.; Dranka, B.; Kumar, S.N.; Myers, C.R.; Bennett, B.; Garces, A.M.; Machado, L.G.D.D.; Thiebaut, D.; Ouari, O.; et al. Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: Potentials, pitfalls, and the future. J. Biol. Chem. 2018, 293, 10363–10380.
- Marques-Aleixo, I.; Santos-Alves, E.; Balça, M.; Rizo-Roca, D.; Moreira, P.; Oliveira, P.; Magalhães, J.; Ascensao, A. Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience 2015, 301, 480–495.
- Wang, D.; Zhai, X.; Chen, P.; Yang, M.; Zhao, J.; Dong, J.; Liu, H. Hippocampal UCP2 is essential for cognition and resistance to anxiety but not required for the benefits of exercise. Neuroscience 2014, 277, 36–44.
- Bristot, V.J.D.O.; Alves, A.C.D.B.; Cardoso, L.R.; Scheffer, D.D.L.; Aguiar, A.S.J. The Role of PGC-1α/UCP2 Signaling in the Beneficial Effects of Physical Exercise on the Brain. Front. Neurosci. 2019, 13, 292.
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950.
- Barbieri, M.; Boccardi, V.; Esposito, A.; Papa, M.; Vestini, F.; Rizzo, M.R.; Paolisso, G. A/ASP/VAL allele combination of IGF1R, IRS2, and UCP2 genes is associated with better metabolic profile, preserved energy expenditure parameters, and low mortality rate in longevity. AGE 2011, 34, 235–245.
- Abdul-Rahman, O.; Sasvari-Szekely, M.; Ver, A.; Rosta, K.; Szasz, B.K.; Kereszturi, E.; Keszler, G. Altered gene expression profiles in the hippocampus and prefrontal cortex of type 2 diabetic rats. BMC Genom. 2012, 13, 81.
- Cardoso, S.; Seiça, R.M.; Moreira, P.I. Uncoupling Protein 2 Inhibition Exacerbates Glucose Fluctuation-Mediated Neuronal Effects. Neurotox. Res. 2017, 33, 388–401.
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, 5314.
- Forsström, S.; Jackson, C.B.; Carroll, C.; Kuronen, M.; Pirinen, E.; Pradhan, S.; Marmyleva, A.; Auranen, M.; Kleine, I.-M.; Khan, N.A.; et al. Fibroblast Growth Factor 21 Drives Dynamics of Local and Systemic Stress Responses in Mitochondrial Myopathy with mtDNA Deletions. Cell Metab. 2019, 30, 1040–1054.e7.
- Basavarajappa, B.; Subbanna, S. Histone Methylation Regulation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4654.
- Muñoz-Carvajal, F.; Sanhueza, M. The Mitochondrial Unfolded Protein Response: A Hinge between Healthy and Pathological Aging. Front. Aging Neurosci. 2020, 12, 300.
- Ren, B.; Zhang, Y.; Zhang, M.; Liu, Y.; Zhang, D.; Gong, X.; Feng, Z.; Tang, J.; Chang, Y.; Zheng, J. Fundamentals of cross-seeding of amyloid proteins: An introduction. J. Mater. Chem. B 2019, 7, 7267–7282.
- Hooper, P.L.; Balogh, G.; Rivas, E.; Kavanagh, K.; Vigh, L. The importance of the cellular stress response in the pathogenesis and treatment of type 2 diabetes. Cell Stress Chaperones 2014, 19, 447–464.
- Kleinridders, A.; Lauritzen, H.P.; Ussar, S.; Christensen, J.H.; Mori, M.; Bross, P.; Kahn, C.R. Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J. Clin. Investig. 2013, 123, 4667–4680.
- Wardelmann, K.; Blümel, S.; Rath, M.; Alfine, E.; Chudoba, C.; Schell, M.; Cai, W.; Hauffe, R.; Warnke, K.; Flore, T.; et al. Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol. Metab. 2019, 21, 68–81.
- Wardelmann, K.; Rath, M.; Castro, J.; Blümel, S.; Schell, M.; Hauffe, R.; Schumacher, F.; Flore, T.; Ritter, K.; Wernitz, A.; et al. Central Acting Hsp10 Regulates Mitochondrial Function, Fatty Acid Metabolism, and Insulin Sensitivity in the Hypothalamus. Antioxidants 2021, 10, 711.
- Chudoba, C.; Wardelmann, K.; Kleinridders, A. Molecular effects of dietary fatty acids on brain insulin action and mitochondrial function. Biol. Chem. 2019, 400, 991–1003.
- Melo, H.M.; Silva, G.D.S.S.D.; Sant’Ana, M.R.; Teixeira, C.V.L.; Clarke, J.R.; Coreixas, V.S.M.; de Melo, B.C.; Fortuna, J.T.; Forny-Germano, L.; Ledo, J.H.; et al. Palmitate Is Increased in the Cerebrospinal Fluid of Humans with Obesity and Induces Memory Impairment in Mice via Pro-inflammatory TNF-α. Cell Rep. 2020, 30, 2180–2194.e8.
- Rosenberger, K.; Dembny, P.; Derkow, K.; Engel, O.; Krüger, C.; Wolf, S.A.; Kettenmann, H.; Schott, E.; Meisel, A.; Lehnardt, S. Intrathecal heat shock protein 60 mediates neurodegeneration and demyelination in the CNS through a TLR4- and MyD88-dependent pathway. Mol. Neurodegener. 2015, 10, 5.
- Juwono, J.; Martinus, R.D. Does Hsp60 Provide a Link between Mitochondrial Stress and Inflammation in Diabetes Mellitus? J. Diabetes Res. 2016, 2016, 8017571.
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588.
- Liyanagamage, D.S.N.K.; Martinus, R.D. Role of Mitochondrial Stress Protein HSP60 in Diabetes-Induced Neuroinflammation. Mediat. Inflamm. 2020, 2020, 8073516.
- Parodi-Rullan, R.; Chapa-Dubocq, X.R.; Javadov, S. Acetylation of Mitochondrial Proteins in the Heart: The Role of SIRT3. Front. Physiol. 2018, 9, 1094.
- Tyagi, A.; Nguyen, C.U.; Chong, T.; Michel, C.R.; Fritz, K.S.; Reisdorph, N.; Knaub, L.; Reusch, J.E.B.; Pugazhenthi, S. SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci. Rep. 2018, 8, 17547.
- Kristian, T.; Karimi, A.J.; Fearnow, A.; Waddell, J.; McKenna, M.C. Perturbed Brain Glucose Metabolism Caused by Absent SIRT3 Activity. Cells 2021, 10, 2348.
- Tyagi, A.; Mirita, C.; Taher, N.; Shah, I.; Moeller, E.; Tyagi, A.; Chong, T.; Pugazhenthi, S. Metabolic syndrome exacerbates amyloid pathology in a comorbid Alzheimer’s mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165849.
- Haigh, J.L.; New, L.E.; Filippi, B.M. Mitochondrial Dynamics in the Brain Are Associated with Feeding, Glucose Homeostasis, and Whole-Body Metabolism. Front. Endocrinol. 2020, 11, 580879.
- Ramírez, S.; Gómez-Valadés, A.G.; Schneeberger, M.; Varela, L.; Haddad-Tóvolli, R.; Altirriba, J.; Noguera, E.; Drougard, A.; Flores, S.R.; Imbernón, M.; et al. Mitochondrial Dynamics Mediated by Mitofusin 1 Is Required for POMC Neuron Glucose-Sensing and Insulin Release Control. Cell Metab. 2017, 25, 1390–1399.e6.
- Huang, S.; Wang, Y.; Gan, X.; Fang, D.; Zhong, C.; Wu, L.; Hu, G.; Sosunov, A.A.; McKhann, G.M.; Yu, H.; et al. Drp1-Mediated Mitochondrial Abnormalities Link to Synaptic Injury in Diabetes Model. Diabetes 2014, 64, 1728–1742.
- Silzer, T.; Barber, R.; Sun, J.; Pathak, G.; Johnson, L.; O’Bryant, S.; Phillips, N. Circulating mitochondrial DNA: New indices of type 2 diabetes-related cognitive impairment in Mexican Americans. PLoS ONE 2019, 14, e0213527.
- Agrawal, R.; Zhuang, Y.; Cummings, B.P.; Stanhope, K.L.; Graham, J.; Havel, P.; Gomez-Pinilla, F. Deterioration of plasticity and metabolic homeostasis in the brain of the UCD-T2DM rat model of naturally occurring type-2 diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1313–1323.