Marine algae are considered to be a potential source of some unique metabolites with diverse health benefits. The pharmacological properties, such as antioxidant, anti-inflammatory, cholesterol homeostasis, protein clearance and anti-amyloidogenic potentials of algal metabolites endorse their protective efficacy against oxidative stress, neuroinflammation, mitochondrial dysfunction, and impaired proteostasis which are known to be implicated in the pathophysiology of neurodegenerative disorders and the associated complications after cerebral ischemia and brain injuries.
- seaweed
- secondary metabolites
- neuroprotection
- Alzheimer’s disease
- Parkinson’s disease
- ischemic stroke
1. Introduction
2. Pathophysiology of Brain Disorders
2.1. Neurodegenerative Disorders (AD and PD)
2.2. Ischemic Stroke
2.3. Traumatic Brain Injury
3. Neuropharmacological Potentials of Marine Algae and Their Metabolites: Evidence from In Vitro Studies
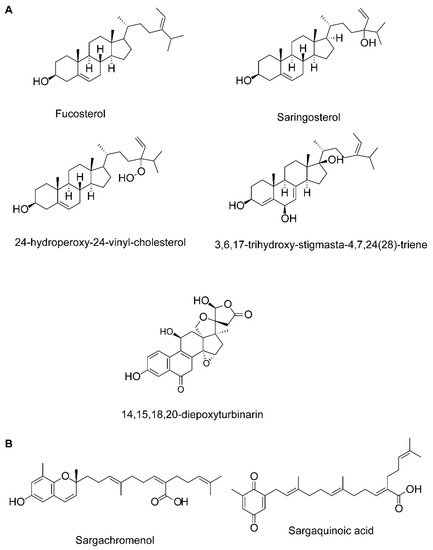
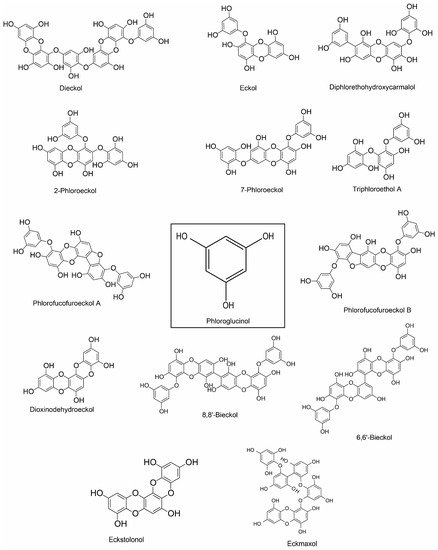
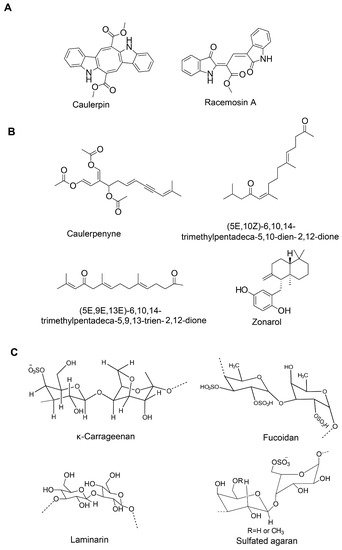
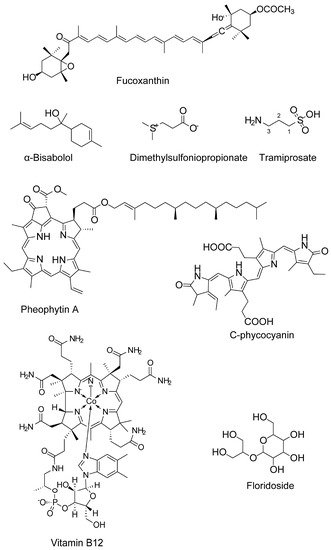
|
Pharmacological Effects |
Compound (Class) |
Algal Source If Any) |
Effective Concentration |
Experimental Model (In Vivo/In Vitro) |
Cellular Effects/Significant Findings |
Signaling Pathways Involved |
Pharmacological Markers |
Reference |
|---|---|---|---|---|---|---|---|---|
|
Antioxidant activity |
Fucoxanthin (carotenoids) |
Sargassum siliquastrum |
50 and 100 μM |
H2O2-induced cell damage in kidney fibroblast cells |
Attenuates oxidative stress |
n.d. |
↓ROS level |
[69] |
|
Fucoxanthin |
5, 10, and 50 μM |
H2O2 induced BV2 microglial cells |
Antioxidation |
Antioxidant pathway |
↓ROS ↑SOD and GSH |
[36] |
||
|
Fucosterol, 3,6,17-trihydroxy-stigmasta-4,7,24(28)-triene and 14,15,18,20-diepoxyturbinarin (sterols) |
Pelvetia siliquosa |
A seven day-dose regimen at 30 mg/kg/day before carbon tetrachloride (CCl4) administration |
Rat model |
Antioxidation |
n.d. |
↑SOD, CAT, and GPx |
||
|
Fucosterol |
Eisenia bicyclis, brown alga |
25, 50, 100, 200, and 400 μM |
RAW 264.7 murine macrophages (t-BHP stimulated) |
Protects against oxidative stress |
n.d. |
↓ROS generation |
||
|
Fucosterol |
Ecklonia stolonifera and Eisenia bicyclis; Brown algae |
25, 50, and 100 μM |
tert-Butyl hydroperoxide- and tacrine-induced HepG2cell injury model |
Antioxidation |
n.d. |
↓ROS generation ↑GSH level |
||
|
Fucosterol |
Sargassum Binderi; brown alga |
3.125, 6.25, 12.5, 25, 50, and 100 μg /mL |
Particulate matter-induced injury and inflammation in A549 human lung epithelial cells |
Attenuates oxidative stress |
↓ROS level ↑SOD, CAT, and HO-1 in the cytosol, and NRF2 in the nucleus |
|||
|
Glycoprotein |
U. pinnatifida |
SOD activity and Xox activity at a concentration of 5 mg/mL and 1 mg/mL, respectively |
In vitro enzyme assay |
↑SOD and↓Xox |
||||
|
Sulfated oligosaccharides |
Ulva lactuca and Enteromorpha prolifera; green algae |
150 mg/kg·day |
Aging model (male senescence-accelerated prone (SAMP8) and male senescence resistant (SAMR1) mice) |
Antioxidantion |
n.d. |
↑GSH, SOD, CAT, telomerase levels, ↑Total antioxidant capacity, ↓MDA and AGEPs |
||
|
Anti-inflammatory activity |
Fucoxanthin |
5, 10, and 50 μM |
Aβ42-induced BV2 microglia cells |
Anti-inflammation |
MAPK pathway |
↓iNOS, COX-2 ↓TNF-α, IL-6, IL-1β, PGE2 ↓JNK, ERK, and p38 MAPK phosphorylation |
[36] |
|
|
Fucoxanthin |
- |
LPS-activated BV-2 microglia |
Anti-inflammation and antioxidation |
Akt/NF-κB and MAPKs/AP-1 pathways; PKA/CREB pathway |
↓iNOS, COX-2, ↓TNF-α, IL-6, PGE2, NO, ROS ↓IL-6, TNF-α, iNOS, and COX-2 mRNA expression ↓Akt, NF-κB, ERK, p38 MAPK and AP-1 phosphorylation ↑Nrf2, HO-1 ↑PKA, CREB ↑BDNF |
|||
|
Fucosterol |
E. bicyclis; brown alga |
5–20 μM for NO |
RAW 264.7 murine macrophages (t-BHP 200 μM, LPS-1μM stimulated) |
↓Inflammatory response |
↓NF-κB pathway |
↓NO production ↓iNOS and COX-2 |
||
|
Fucosterol |
U. pinnatifida |
10, 25, or 50 μM |
LPS-induced RAW 264.7 macrophages and THP-1 human monocyte cell line |
↓Inflammatory response |
↓NF-κB pathway |
↓iNOS, TNF-α, and IL-6 ↓DNA binding ↓phosphorylation of NF-κB, MKK3/6 and MK2 |
||
|
Fucosterol |
Hizikia fusiformis |
1–10 μM |
CoCl2 induced hypoxia in keratinocytes |
↓Inflammatory response |
n.d. |
↓IL-6, IL-1β and TNF-α ↓pPI3K and pAkt and HIF1-α accumulation |
||
|
Fucosterol |
Panida. australis |
0.004,0.2, and 10 μM |
LPS or Aβ-induced BV2 (microglial) cells |
Protects against LPS or Aβ-mediated neuroinflammation |
n.d. |
↓IL-6, IL-1β, TNF-α, NO, and PGE2 |
||
|
Fucosterol |
S. Binderi; brown alga |
3.125, 6.25, 12.5, 25, 50, 100 μg/mL |
Particulate matter-induced injury and inflammation in A549 human lung epithelial cells |
↓Inflammatory response |
n.d. |
↓COX-2, PGE2, TNF-α and IL-6 |
||
|
Dieckol (phlorotannin) |
E. cava |
50–300 µg/mL |
LPS-stimulated murine BV2 microglia |
Anti-inflammation and antioxidation |
p-38 MAPK/ NF-κB pathway |
↓NO and PGE2; ↓iNOS and COX-2; ↓IL-1β and TNF-α; ↓ROS |
||
|
Phloroglucinol, eckol, dieckol, 7-phloroeckol, phlorofucofuroeckol A and dioxinodehydroeckol (phlorotannin) |
E. bicyclis; brown alga |
5–20 μM for NO |
LPS-stimulated RAW 264.7 murine macrophages |
↓Inflammatory response |
↓NF-κB pathway |
↓NO production |
||
|
Phlorofucofuroeckol A |
E. stolonifera |
20 μM |
LPS-activated BV2 and primary microglial cells |
Anti-inflammation |
NF-κB, JNKs, p38 MAPK, and Akt pathways |
↓NO and PGE2; ↓iNOS and COX-2; ↓IL-1β, IL-6 and TNF-α; ↓NF-κB activation and IκB-α degradation ↓JNK, p38, and Akt |
||
|
Phlorofucofuroeckol B (phlorotannin) |
E. stolonifera |
10–40 µM |
LPS-stimulated murine BV2 microglia |
Anti-inflammation |
IκB-α/NF-κB and Akt/ERK/JNK pathways |
↓TNF-α, IL-1β and IL-6; ↓COX-2 and iNOS ↓NF-κB activation and IκB-α degradation ↓Akt, ERK, and JNK phosphorylation |
||
|
8,8’-bieckol (phlorotannin) |
E. cava |
LPS-stimulated primary macrophages and RAW 264.7 macrophages & LPS-induced septic mice |
Anti-inflammation; Protects mice from endotoxin shock |
NF-κB pathway |
↓NO and PGE2; ↓iNOS mRNA and protein expression; ↓IL-6; ↓Transactivation of NF-κB and nuclear translocation of the NF-κB p65 subunit ↓ROS |
|||
|
6,6′-bieckol (phlorotannin) |
E.stolonifera |
LPS-stimulated BV2 and murine primary microglial cells |
Anti-inflammation |
IκB-α/NF-κB and JNK/p38 MAPK/Akt pathways |
↓COX-2 and iNOS; ↓NO and PGE2, ↓IL-6 ↓Transactivation of NF-κB and nuclear translocation of the NF-κB p65 subunit ↓Akt, JNK and p38 MAPK phosphorylation |
|||
|
Fucoidan (sulfated polysaccharide) |
Brown seaweed |
25, 50, and 100 µg/mL |
LPS-stimulated murine BV2 microglia |
Anti-inflammation |
NF-κB and JNK/p38 MAPK/Akt pathways |
↓NO and PGE2; ↓COX-2, iNOS and MCP-1; ↓TNF-α and IL-1β; ↓NF-κB activation; ↓Akt, ERK, p38 MAPK and JNK phosphorylation |
||
|
Fucoidan |
- |
125 µg/mL |
LPS-activated primary microglia |
Anti-inflammation |
n.d. |
↓TNF-α and ROS |
||
|
κ-carrageenan oligosaccharides and desulfated derivatives |
Red algae |
LPS-activated microglia |
Anti-inflammation |
n.d. |
↓TNF-α |
|||
|
Sulfated oligosaccharides |
U. lactuca and E. prolifera; green algae |
150 mg/kg·day |
Aging model (male senescence-accelerated prone (SAMP8) and male senescence resistant (SAMR1) mice) |
↓Inflammatory response |
n.d. |
↓IFN-γ, TNF-α, and IL-6 |
||
|
Alginate-derived oligosaccharide |
Brown algae |
50–500 µg/mL |
LPS/Aβ-stimulated BV2 microglia |
Anti-inflammation |
TLR4/NF-κB signaling pathway |
↓NO and PGE2; ↓COX-2 and iNOS; ↓TNF-α, IL-6 and IL-12; ↓TLR4; ↑NF-κB/p65 subunit translocation |
||
|
Seleno-polymannuronate |
Brown algae |
0.8 mg/mL |
LPS-activated primary microglia and astrocytes; mouse model of acute inflammation |
Anti-inflammation |
NF-κB and MAPK signaling |
↓NO and PGE2; ↓COX-2 and iNOS; ↓TNF-α, IL-1β and IL-6; ↑IκB-α, p65, p38, ERK and JNK phosphorylation |
||
|
Sargachromenol (plastoquinone) |
Sargassum micracanthum |
30.2 μM (IC50) |
LPS-stimulated RAW 264.7 macrophages |
Anti-inflammation |
NF-κB signaling |
↓NO and PGE2; ↓COX-2 and iNOS; ↑IκB-α |
||
|
Sargaquinoic acid (plastoquinone) |
Sargassum siliquastrum |
LPS-stimulated RAW 264.7 macrophages |
Anti-inflammation |
NF-κB signaling |
↓NO; ↓iNOS; ↑IκB-α; ↓nuclear translocation of NF-κB; ↓JNK1/2 MAPK |
|||
|
Floridoside (glycerol glycosides) |
Laurencia undulate; red alga |
50 μM |
LPS-stimulated murine BV2 microglia |
Anti-inflammation |
MAPK Signaling |
↓NO, ROS; ↓iNOS and COX-2; ↓p38 MAPK and ERK phosphorylation |
||
|
Glycoprotein |
U. pinnatifida |
COX-1 and COX-2 inhibition with IC50 values of 53.03 ± 1.03 μg/mL and 193.35 ± 3.08 μg/mL, respectively |
LPS-stimulated RAW 264.7 macrophages |
Anti-inflammation |
n.d. |
↓COX-1 and COX-2 ↓NO |
||
|
Caulerpin (bisindole alkaloid) |
Caulerpa racemosa |
100 µM/kg body wt |
Capsaicin-induced ear edema and carrageenan-induced peritonitis |
Inhibition of nociception |
n.d. |
n.d. |
||
|
Caulerpenyne (sesquiterpene) |
C. prolifera and C. racemosa |
5.1 μM |
Lipoxygenase (LOX) enzyme activity assay |
Inhibitory activity against LOX |
- |
Un-competitive type of inhibition |
||
|
Aquamin (multi-mineral complex) |
Lithothamnion corallioides; red alga |
LPS-stimulated, glial-enriched primary cultures of rat cortex |
Anti-inflammation |
n.d. |
↓TNF-α and IL-1β |
|||
|
Anticholinesterase activity |
Fucosterol and 24-hydroperoxy 24-vinylcholesterol |
E. stolonifera |
IC50 values of 421.72 ± 1.43, 176.46 ± 2.51 µM, respectively |
In vitro enzymatic assay |
↓BChE activity |
- |
Selective inhibition of BChE |
|
|
Fucosterol |
Panida australis |
inhibition against AChE (10.99–20.71%) and BChE (4.53–17.53%) with concentrations ≤ 56 μM, |
In vitro enzymatic assay |
↓AChE and BChE activities |
- |
Nonselective cholinesterase inhibition |
||
|
Fucosterol |
Sargassum horridum |
- |
In vitro enzymatic assay |
↓AChE activity |
- |
Non-competitive inhibition |
||
|
Fucoxanthin |
- |
IC50 value 1.97 mM |
In vitro BChE activity assay |
↓BChE activity |
Mixed inhibition type |
|||
|
Fucoxanthin |
Brown seaweed |
IC50 value of 81.2 μM |
In vitro AChE activity assay; Molecular docking analysis |
↓AChE activity |
Fucoxanthin likely interacts with the peripheral anionic site within AChE |
Non-competitive manner |
||
|
α-Bisabolol |
Padina gymnospora |
IC50 value < 10 μg/mL |
In vitro enzymatic assay |
↓AChE and BChE activity |
- |
- |
||
|
Glycoprotein |
U. pinnatifida |
AChE and BChE inhibitory activities with IC50 values of 63.56 ± 1.86 and 99.03 ± 4.64, respectively |
In vitro enzymatic assay |
↓AChE and BChE activity |
- |
- |
||
|
Phloroglucinol, |
Ecklonia maxima; Brown alga |
IC50 value: 76.70 to 579.32 μM |
In vitro AChE activity assay |
↓AChE activity |
- |
- |
||
|
Dieckol and phlorofucofuroeckol |
E. cava |
Ethanol-intoxicated memory impairment in mice |
↓AChE activity |
n.d. |
↑Acetylcholine |
|||
|
Sargaquinoic acid and sargachromenol (plastoquinones) |
Sargassum sagamianum |
IC50 value for anti-AChE: 23.2 and 32.7 μM, respectively; IC50 value for anti-BChE of sargaquinoic acid 26 nm |
In vitro ChE activity assay |
Sargaquinoic acid shows potent inhibitory activity against BuChE and moderate inhibitory activity against AChE |
-. |
- |
||
|
(5E,10Z)-6,10,14-trimethylpentadeca-5,10-dien-2,12-dione and (5E,9E,13E)-6,10,14-trimethylpentadeca-5,9,13-trien-2,12-dione (Sesquiterpenes) |
S. sagamianum |
IC50 values of 65.0 and 48.0, and 34.0 and 23.0 μM, respectively |
In vitro ChE activity assay |
Moderate inhibitory activity against AChE and BuChE |
- |
- |
||
|
Anti-amyloidogenic and aggregation inhibition activity |
Fucoxanthin |
E. stolonifera and U. pinnatifida |
↓β-secretase activity; Binding energy (-7.0 kcal/mol) |
- |
mixed-type inhibition |
|||
|
Fucoxanthin |
- |
0.1–30 μM |
Suppresses the formation of Aβ1-42 fibrils and Aβ1–42 oligomers, and inhibits Aβ aggregation |
- |
- |
|||
|
Fucoxanthin |
- |
2 μM |
ThT assay |
Inhibits Aβ1-42 fibril and aggregate formation |
- |
- |
||
|
Fucosterol |
E. stolonifera and U. pinnatifida |
10–100 μM (IC50 value of 64.12 ± 1.0 μM) |
In vitro enzyme assay; In silico analysis |
↓β-secretase activity; Binding energy (−10.1 kcal/mol) |
- |
Noncompetitive inhibition |
||
|
α-Bisabolol |
Padina gymnospora |
5 μg/mL |
Thioflavin T (ThT), Confocal laser scanning microscopy (CLSM) analysis, Transmission electron microscopy (TEM), Fourier transform infrared (FTIR) spectroscopic analysis and molecular dynamics simulation |
Prevents oligomers formation as well as disaggregates the matured fibrils |
- |
- |
||
|
Glycoprotein |
U. pinnatifida |
IC50 values of 73.35 ± 2.54 μg/mL |
In vitro enzymatic assay |
↓BACE1 activity |
- |
- |
||
|
Cholesterol homeostasis and Aβ clearance activity |
Fucosterol |
- |
100 and 200 μM (HEK293 cell cultures); 100 or 200 μM (macrophages and HepG2, H4IIE, and Caco2 cells) |
HEK293 cell cultures (Reporter system); THP-1-derived macrophages; Caco-2 cells HepG2 cells |
Reverses cholesterol transport. No accumulation of triglyceride in HepG2 |
n.d. |
Dual-LXR agonist (LXR-α and LXR-β) ↑ABCA1, ABCG1, and ApoE; ↑Intestinal NPC1L1 and ABCA1; ↑Insig-2a, that delays nuclear translocation of SREBP-1c |
|
|
Saringosterol |
Sargassum fusiforme |
30 μM |
Luciferase reporter assay system; HEK293T, THP-1 monocytes, HepG2, RAW264.7, THP-1 macrophages and Caco-2 cells |
n.d. |
n.d. |
Selective LXRβ agonist; ↑ABCA1, ABCG1, and SREBP-1c |
||
|
Alginate-derived oligosaccharide |
Marine brown algae |
BV2 microglial cells |
Microglial phagocytosis of Aβ |
Toll-like receptor signaling |
↑TLR4 |
|||
|
Monoamine oxidase inhibition and affinity to dopaminergic receptors |
Phlorofucofuroeckol-A and dieckol (phlorotannin) |
- |
In vitro enzyme assay and functional assay for GPCR screening; Docking analysis |
↓hMAO activity; D3R and D4R stimulation |
- |
- |
||
|
Antiaging |
Sulfated oligosaccharides |
U. lactuca and E. prolifera; green algae |
150 mg/kg/day |
Aging model (male senescence-accelerated prone (SAMP8) and male senescence resistant (SAMR1) mice) |
Antioxidant and anti-inflammation |
n.d. |
↑GSH, SOD, CAT, telomerase levels, ↑Total antioxidant capacity, ↓MDA and AGEPs ↓IFN-γ, TNF-α, and IL-6 ↑BDNF and ChAT; ↑Sirt1, ↑p53 and FOXO1 |
|
|
Fucosterol |
Hizikia fusiformis |
50 µg/mL |
Culture model of C. elegans |
Extends lifespan |
↑Antioxidant mechanism |
n.d. |
n.d.: not defined; -: information not available.
4. Neuropharmacological Potentials of Marine Algae and Their Metabolites: Evidence from In Vivo Studies
5. Conclusion and Future perspectives
Although neuroactive compounds were isolated from a range of algae, seaweed species under Phaeophyceae yield the highest number of compounds. However, species from other groups, for example, Gelidium amansii under Rhodophyceae that exhibited significant neuromodulatory effects, also could offer some promising metabolites. Moreover, a large number of species remain unexplored. While degenerating brains experience disruption of synaptic connectivity, compounds with neuritogenic capacity may potentially enhance the regeneration of damaged processes. Therefore, compounds, both neuroprotective and neurotrophic, are equally important. However, in contrast to neuroprotective compounds that potentially support neuronal survival, a few compounds showing neurite outgrowth potential have been discovered in marine algae. Compounds, including those that have already shown neuroprotective ability as well as those that have not yet been explored, therefore, need to be screened for their ability to promote neurite extension.
Despite a sizable collection of algae-based natural products with distinct neuroprotective functions, only sodium oligomannate has emerged as a successful drug for AD. This work, therefore, calls for intensive research on other potential compounds to translate the preclinical findings into clinical models. In addition, the factors that are responsible for the failure of a clinical trial need to be carefully reviewed. For example, the bioavailability of a candidate drug in the brain, including its ability to cross BBB, remains one of the barriers to therapeutic success. If the ADME (absorption, distribution, metabolism, and excretion) properties of a preclinically effective compound sufficiently guarantee its drug-likeliness, it is highly likely that the compound may succeed in clinical trials. This is why the ongoing strategy requires a rational reformation incorporating modern approaches, such as virtual screening and system biology, to strengthen the algae-based drug development process. The computational study will provide some crucial information on the ADME properties of potential leads and its interaction and binding affinity to molecular targets while system biology knowledge will identify the potential interaction of target molecules and cellular signaling pathways at the systemic level. With the constant discovery of new compounds, all these strategies will accelerate the designing and development of algae-based future drugs.Despite a sizable collection of algae-based natural products with distinct neuroprotective functions, only sodium oligomannate has emerged as a successful drug for AD. This review, therefore, calls for intensive research on other potential compounds to translate the preclinical findings into clinical models. In addition, the factors that are responsible for the failure of a clinical trial need to be carefully reviewed. For example, the bioavailability of a candidate drug in the brain, including its ability to cross BBB, remains one of the barriers to therapeutic success. If the ADME (absorption, distribution, metabolism, and excretion) properties of a preclinically effective compound sufficiently guarantee its drug-likeliness, it is highly likely that the compound may succeed in clinical trials. This is why the ongoing strategy requires a rational reformation incorporating modern approaches, such as virtual screening and system biology, to strengthen the algae-based drug development process. The computational study will provide some crucial information on the ADME properties of potential leads and its interaction and binding affinity to molecular targets while system biology knowledge will identify the potential interaction of target molecules and cellular signaling pathways at the systemic level. With the constant discovery of new compounds, all these strategies will accelerate the designing and development of algae-based future drugs.