Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Nicolas MASURIER.
Thienopyrimidine emerges as an attractive scaffold in medicinal chemistry with a wide array of pharmacological properties, such as antibacterial, antifungal, antiparasitic and antiviral. Considering the fusion between pyrimidine and thiophene rings, three different thienopyrimidines can be obtained, namely thieno[2,3-d]pyrimidines, thieno[3,2-d]pyrimidines and thieno[3,4-d]pyrimidines. Different synthetic pathways involving the construction of the pyrimidine or the thiophene ring were reported in the literature to access polysubstituted thienopyrimidines. In these approaches, the synthetic strategies mostly involved the synthesis of a thienopyrimidin-4-one derivative, where position 4 could be modified via further functionalization.
- thienopyrimidine
- heterocycle
- cyclization
- synthesis
1. Synthesis from Thiophene Derivatives
Due to the high diversity of supplies, the reaction between an aminothiophene derivative bearing an electrophilic center (ester or nitrile) and a carbonyl or an amine reactant is probably the easiest way for produce thienopyrimidin-4-one derivatives. The leading routes to afford thienopyrimidines from aminothiophene derivatives are described in Scheme 1.
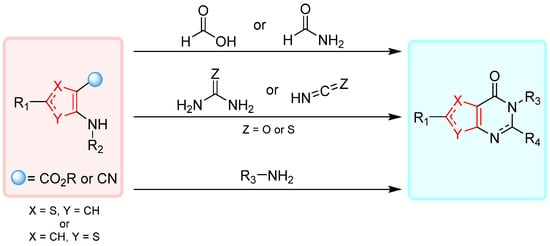
Scheme 1.
Main synthetic pathways to produce thienopyrimidin-4-ones from thiophene derivatives.
1.1. Cyclization with Carbonyl Reactants
The most efficient chemical approach to access 2- and 3-unsubstituted thieno[2,3-d]pyrimidin-4(3H)-ones involved a condensation reaction between an aminothiophene substrate and formamide. Thus, compounds 1a–e treated with an excess of formamide at high temperature led to compounds 2a–e with good yields (76 to 97%), except for compound 1e for which the methoxy group in R3 decreased the reaction yields compared to the ethoxy group (1a) (Scheme 2) [10,11,12,13,14][1][2][3][4][5].
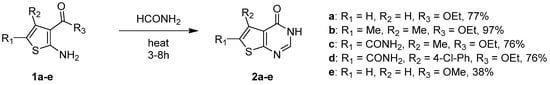
Scheme 2. Access to 2- and 3-unsubstituted thieno[2,3-d]pyrimidin-4-one derivatives (Me = methyl, Et = ethyl, and Ph = phenyl).
In contrast, mild conditions were sufficient to perform cyclization reaction with formamide to synthesize the thieno[3,2-d]pyrimidin-4(3H)-one isomers 4a–b with good yields (60 to 65%, Scheme 3) [15][6].
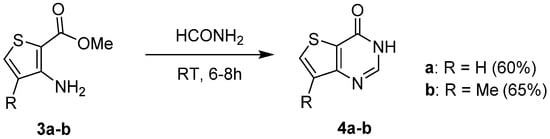
Scheme 3.
Synthesis of 3-unsubstituted thieno[3,2-
d
]pyrimidines
4a–b
.
Woodring et al. presented a variant of this process that also involved formamide in combination with ammonium formate [14][5]. Cyclization of the thiophene intermediate 5 at 150 °C led to the unsubstituted thieno[3,2-d]pyrimidin-4-one 6 with a 56% yield (Scheme 4).
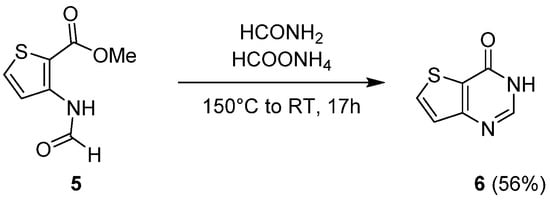
Scheme 4.
Synthetic route to unsubstituted thieno[3,2-
d
]pyrimidin-4-one
6
.
In addition, reaction of 2-amino-3-cyanothiophene derivatives with formic acid could also be considered to access 2- and 3-unsubstituted thieno[2,3-d]pyrimidin-4-ones [13][4]. In such approach, the cyano group is firstly converted into its corresponding primary amide, which could then be cyclized in the presence of formic acid. Kanawade et al. used such an approach to prepare thienopyrimidinone 8a from 2-amino-3,5-dicyanothiophene 7a (Scheme 5). Replacing formic acid by formamide led to the formation of the 4-amino analogue, as reported by Aly et al. [16][7]. Thus, cyclocondensation involving 7b and formamide occurred under reflux to afford the expected 8b with a 83% yield (Scheme 5).
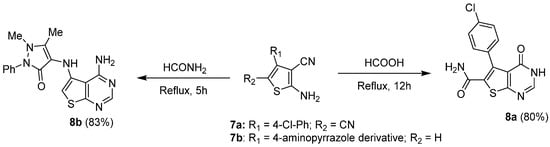
Scheme 5.
Access to thieno[2,3-
d
]pyrimidine derivatives from 2-amino-3-cyanothiophene derivatives.
Cyclocondensation of thiophene carboxamide 9 in the presence of sodium hydroxide was used to synthesize thieno[3,4-d]pyrimidin-4(3H)-one 10 (Scheme 6). The expected molecule was isolated with a moderate yield (40%) after a 1 h reaction in refluxing methanol.
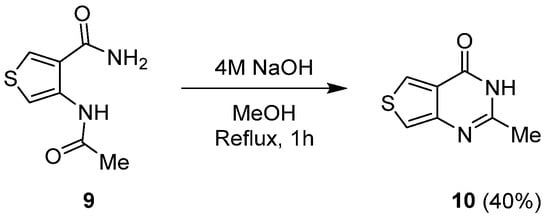
Scheme 6.
Synthesis of 2-methyl-thieno[3,4-
d
]pyrimidin-4(3
H
)-one
10
.
Using a similar approach, but with a nitrile group as the precursor of the primary amide, Desroches et al. synthesized 2-methyl- and 2-trichloromethyl-thieno[2,3-d]pyrimidin-4(3H)-ones 12 and 13, respectively (Scheme 7) [17][8]. Thus, treatment of 3-cyanothiophene acetamide 11a with hydrogen peroxide in alkaline medium (NaOH) afforded 2-methyl-thieno[2,3-d]pyrimidin-4(3H)-one 12 with a 72% yield. Using 3-cyanothiophene trichloroacetamide as a substrate and phosphoric acid in polyphosphoric acid triggered the cyclocondensation reaction and the formation of the 2-trichloromethyl-thieno[2,3-d]pyrimidin-4(3H)-one 13 with good yields (90%).
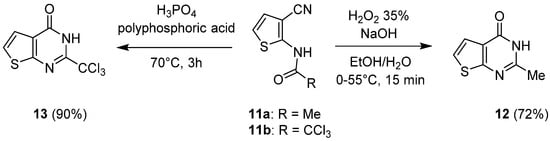
Scheme 7.
Synthesis of thieno[2,3-
d
]pyrimidin-4(3
H
)-ones substituted in position 2.
1.2. Cyclization with Nitrile Reactants
Various pathways exploiting nitrile condensation were reported in the literature to produce thieno-fused analogues. De Schutter et al. used a synthetic route involving a thiophene amino ester treated in strongly acidic conditions by a cyanoalkyl derivative at 90 °C (Scheme 8) [18][9]. Thieno[2,3-d]pyrimidin-4(3H)-ones 16c, substituted in positions 2, 5, and 6 were then obtained in 1,4-dioxane in moderate to good yields (50 to 90%). In addition, Mavrora et al. used the same synthetic pathway and obtained chloroethyl derivatives 16a–b with good yields (Scheme 8) after nitrile cyclocondensation at room temperature [19][10]. Likewise, thieno[3,2-d]pyrimidinones 15 substituted at position 2 were prepared from cyclization of the starting thiophene with the appropriate cyanoalkyl in acidic conditions at 90 °C in 1,4-dioxane (Scheme 8) [18][9]. To introduce a trichloromethyl group at position 2 of the thieno[3,2-d]pyrimidine core, Desroches et al. used trichloroacetonitrile in acetic acid, saturated with HCl gas, to afford 2-trichloromethyl-thieno[3,2-d]pyrimidine 17 with a 63% yield (Scheme 8) [17][8].
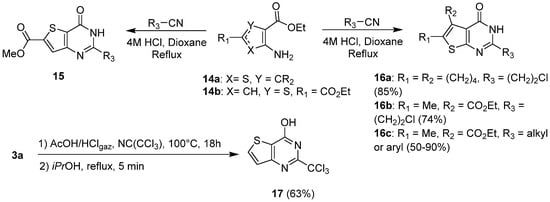
Scheme 8.
Synthesis of 2-substituted thienopyrimidin-4-ones using nitrile reactants.
Using the same strategy, Kim et al. introduced a chloromethyl group at position 2 of thieno[3,2-d]pyrimidinones after slight modifications of the reaction conditions [20][11]. Formation of the thieno-fused core occurred with the cyclocondensation of malononitrile with 2-methyl-3-aminothiophene carboxylate under acidic conditions and mild heating to offer 18 with high yields (Scheme 9).
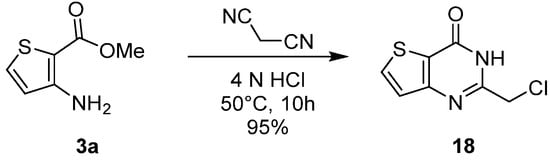
Scheme 9.
Synthesis of 2-chloromethyl-thieno[3,2-
d
]pyrimidinone
18
.
Slavinski et al. presented another synthetic pathway to introduce a sulfonamide group at position 2, using sulfonyl cyanamide potassium salts 19 [21][12]. Acidification of the reaction with boiling glacial acetic acid led to cyclization and afforded 2-sulfonamide-thieno[3,2-d]pyrimidinone derivatives 20 with low yields (20–34%, Scheme 10).
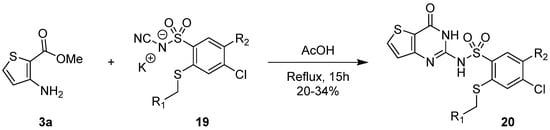
Scheme 10.
Formation of 2-sulfonamide-thieno[3,2-
d
]pyrimidinones
20
.
1.3. Synthesis from (Thio)urea Reagents, Iso(Thio)cyanate or (Thio)cyanate Derivatives
An easy way to access thienopyrimidin-2,4-dione or 2-thioxo-thienopyrimidin-4-one derivatives consisted of cyclocondensation of the appropriate ethyl aminothiophene-carboxylate with potassium (thio)cyanate in an acidic medium. Patel et al. obtained 2-thioxo-thieno[2,3-d]pyrimidin-4-one 22a with a 58% yield, using hydrochloric acid in refluxing 1,4-dioxane (Scheme 11) [22][13], whereas Temburkinar et al. and other groups [23,24,25][14][15][16] used potassium cyanate in acetic acid to obtain thieno[3,2-d]pyrimidin-2,4-dione 21a with 71 to a 88% yield.
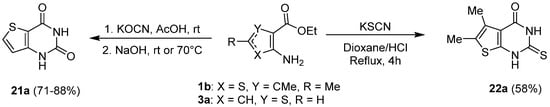
Scheme 11.
Synthesis of 2-thioxo-thieno[2,3-
d
]pyrimidin-4-one
22a
and thieno[3,2-
d
]pyrimidin-2,4-dione
21a
.
Another way to access such compounds was to condensate the starting aminothiophene with urea or thiourea, followed by cyclization to afford thienopyrimidinone compounds 21 or 22. Ortikov and Prabhakar teams used such conditions to synthesize 2-thioxo-thieno[2,3-d]pyrimidin-4-one 22b and thieno[2,3-d]pyrimidine-2,4-diones 22c (Scheme 12) with good yields (72–91%) [11,26,27][2][17][18]. Condensation and cyclization only occurred at very high temperatures after 2 or 3 h of heating without solvent. Thieno[3,2-d]pyrimidin-2,4-one 21b could be synthesized under these conditions, whereas the synthesis of 2-thioxo-thieno[3,2-d]pyrimidin-4-ones 21c required the use of N,N-dimethylformamide (DMF) as a solvent (Scheme 12) [28,29][19][20].
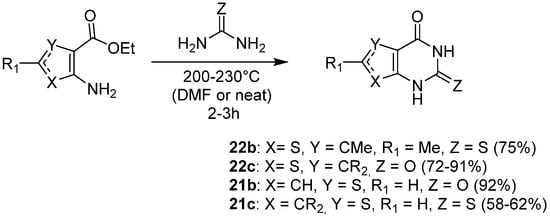
Scheme 12.
Formation of 2-thioxo-thienopyrimidin-4-ones and thienopyrimidine-2,4-diones using (thio)urea.
Kankanala et al. used a common synthetic pathway to access 3-hydroxythieno[2,3-d]pyrimidin-2,4-diones and thieno[3,2-d]pyrimidin-2,4-diones [30][21] bearing various groups in α and β positions of the sulfur atom. Firstly, the aminothiophene reacted with 1,1′-carbonyldiimidazole (CDI) to afford the imidazole-carboxamide intermediate after 2 h in refluxing toluene (Scheme 13). Secondly, the substitution of the imidazole group by protected hydroxylamine generated the hydroxyurea intermediate. Then, a basic treatment deprotonated hydroxyurea to allow cyclization. Afterward, deprotection of the hydroxyurea led to the final compounds 23 with correct to good yields (40–85%).
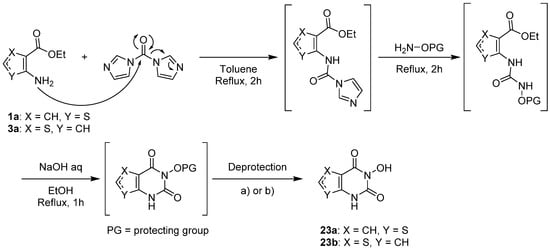
Scheme 13.
Synthetic pathway to afford 3-hydroxythienopyrimidin-2,4-diones
23
.
To introduce more chemical diversity at position 3, a convenient synthetic route described by Abu-Hashem et al. involved nucleophilic attack of an aminothiophene derivative on an isocyanate or thioisocyanate in the presence of a catalytic amount of triethylamine in refluxing 1,4-dioxane (Scheme 14A) [31][22]. The (thio)ureidothiophene intermediate 24 or 25 was then isolated on average with good yields (60 and 70%). Thereafter, basic treatment of 24 or 25 with sodium ethoxide in refluxing ethanol led to thieno-fused derivatives 26 and 27 with good yields (70% and 75%) after 8 h. Dewal et al. obtained similar results using sodium methoxide under refluxing methanol to prepare trisubstituted thieno[2,3-d]pyrimidin-2,4-dione derivatives 26 with 88–90% yields [32][23]. In addition, Abu-Hashem et al. reported a one-pot reaction with phenylisothiocyanate and sodium hydroxide as a base, in refluxing ethanol for 6 h [31][22]. Both the two-step procedure and the one-pot reaction offered 27a with a 70% yield (Scheme 14A). Furthermore, the use of potassium carbonate in refluxing acetonitrile led to the 2-mercapto-thieno[2,3-d]pyrimidin-4-one analogues 28b–c in even higher yields (78%) [12,33][3][24]. In a similar way, 3-ethyl-2-thioxo-thieno[3,2-d]pyrimidin-4-one 27b was also accessible via the cyclization of 2-methyl-3-aminothiophene carboxylate with ethylisothiocyanate in refluxing pyridine [34][25]. In addition, 6-bromothieno[3,2-d]pyrimidin-2,4-diol 30 was synthesized in milder conditions with potassium tert-butoxide in DMF at room temperature and obtained it with a quantitative yield (Scheme 14B) [35][26]. It was then possible to introduce further chemical diversity in positions 2, 4, and 6, starting from this bicyclic product.
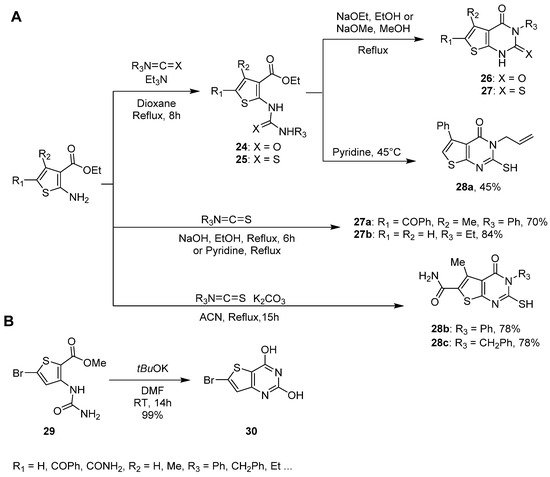
Scheme 14. (A). Synthesis of 3-substituted 2-thioxo-thienopyrimidin-4-ones or thienopyrimidine-2,4-diones 26–28. (B). Synthesis of 6-bromothieno[3,2-d]pyrimidine-2,4-diol 30.
Alternately, Cohen et al. suggested an original synthetic pathway to obtain thieno[3,2-d]pyrimidin-4(3H)-one derivatives 34, substituted in position 2 by an amino group [36][27]. This one-pot procedure involved first the condensation of the starting material with ethoxycarbonyl isothiocyanate in DMF to generate the thiourea carbamate intermediate 32, that was not isolated (Scheme 15). Afterward, a primary alkylamine reacted with this species, previously mixed with 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide (EDCI.HCl) and triethylamine. Guanidine intermediate 33 was observed but was not isolated. Then, this intermediate cyclized at 170 °C to afford thieno-fused derivatives 34 with 42 to 70% yields depending on the substituents.
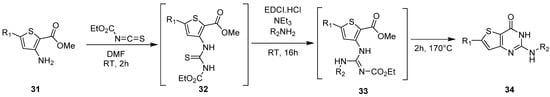
1.4. Synthesis via a Tetrazole Intermediate
To generate thieno[2,3-d]pyrimidines substituted in positions 2 and 3 by an amino group, Abu-Hashem et al. purposed an access route via a tetrazole intermediate (Scheme 16) [31][22]. Firstly, the tetrazole ring was formed by treating 35 with triethyl orthoformate and sodium azide to generate 36 with good yields (70%). Then, refluxing 36 in the presence of a large excess of hydrazine hydrate led to two consecutive hydrazide intermediates 37 and 38. Intramolecular cyclization of 38 afforded 39 with good yields (75%).
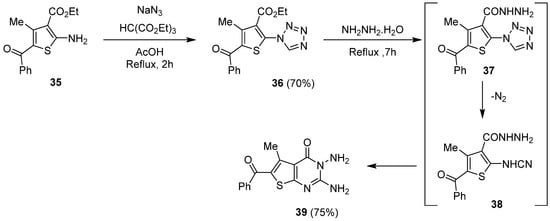
Scheme 16.
Synthesis of 2,3-diaminothieno[2,3-
d
]pyrimidine
39
.
1.5. Cyclization with Amine/Hydrazine Derivatives
A more common way to access 3-amino-thieno[2,3-d]pyrimidin-4-ones consisted of the condensation and cyclization between a thiophene derivative and hydrazine monohydrate in refluxing ethanol. Using this strategy, several groups reported the synthesis of compounds 42a–b with moderate to good yields (Scheme 17) [12,37][3][28]. Aly et al. employed the same reaction conditions to generate 3-amino-thieno[2,3-d]pyrimidin-4-one 42c. Only the starting thiophene was different and achieved cyclocondensation with good yields (80%).
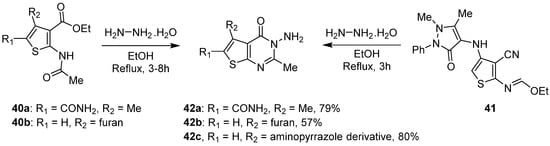
Scheme 17.
Synthesis of 3-amino-thienopyrimidin-4-ones
42
.
To introduce chemical diversity at position 3, a similar route was followed by Habib et al. using various primary amines to synthesize a set of 3-substituted thieno[2,3-d]pyrimidinone derivatives 43 [12][3]. Firstly, the 2-aminothiophene 1c reacted with triethyl orthoformate under reflux to prepare the imino intermediate, which was not isolated (Scheme 18). Then, the appropriate amine was added to allow cyclization and obtain 3-substituted thienopyrimidinone derivatives 43 with good yields (79–85%).
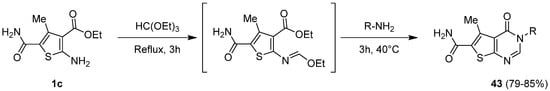
Scheme 18.
Access route to synthesize 3-substituted thieno[2,3-
d
]pyrimidin-4-ones
43
.
Finally, condensation of ammonia with N-acylaminothiophenes 44 allowed access to 3-unsubstituted thieno[2,3-d]pyrimidin-4-ones 45 [15,17][6][8]. The first synthetic route involved 25% ammonia heated at 105 °C in a sealed vial to obtain thieno[3,2-d]pyrimidin-4-one 46a after 3 h, with a 63% yield (Scheme 19). In contrast, using milder conditions with 30% ammonia at room temperature for 6 to 8 h led generally to lower yields (28–60%). Moreover, it has been observed by Desroches et al. that this method was not efficient when R = CCl3 (compound 45e) [17][8]. Indeed, with this substrate, cyclization in the presence of 25% ammonium hydroxide in a sealed vial failed.
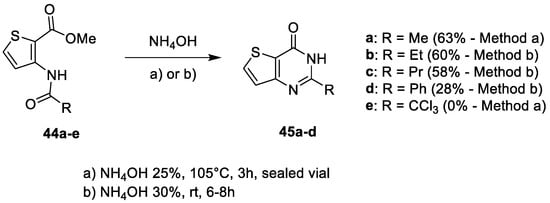
Scheme 19.
Synthesis of 3-unsubstituted-thienopyrimidin-4-ones
45
(Pr = propyl).
2. Synthesis of Thienopyrimidines from Pyrimidine Derivatives
2.1. Synthesis from the Thorpe-Ziegler Reaction
One of the possibilities to shape the thieno-fused ring from pyrimidine derivatives is the Thorpe-Ziegler cyclization. A six-membered ring bearing a mercaptocarbonitrile group was the starting point to synthesize thienopyrimidines (Scheme 20). After substitution of alkyl chloroacetate by the sulfhydryl group (compound 47), and subsequent deprotonation, cyclization can occur in basic conditions. In such a way, Abdel Hamid et al. reported the synthesis of compound 48 with a 71% yield [38][29].
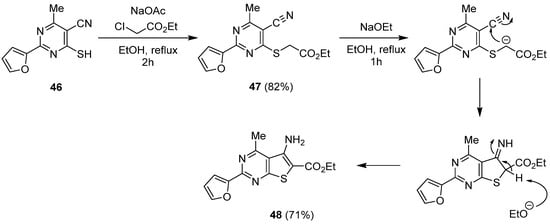
Scheme 20.
Synthesis of thienopyrimidin-4-one
48
via a Thorpe-Ziegler cyclization.
A variant of the previous approach was purposed by Ali and Saleh for the synthesis of 2-thioxo-1,2,3,4-tetrahydrothieno[3,4-d]pyrimidine 52 [39][30]. First, thiobarbituric acid 49 was deprotonated in α-position of the two carbonyl groups at room temperature (Scheme 21). Then, nucleophilic substitution on phenyl isothiocyanate led to the ketene aminothioacetal 50. Thereafter, the addition of alkyl bromoacetate allowed cyclocondensation of 51 in basic conditions. The final product 52 was obtained with good yields (74%).
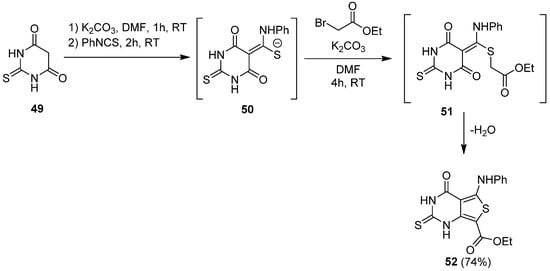
Scheme 21.
Synthesis of 2-thioxo-1,2,3,4-tetrahydro thieno[3,4-
d
]pyrimidin-4-one
52
.
2.2. Synthesis from the Gewald Reaction
The Gewald reaction is a versatile reaction to access 2-aminothiophene derivatives involving one-pot cyclocondensation of ketones or aldehydes with activated nitrile derivatives and elemental sulfur. Using thiobarbituric acid 49 as the starting ketone, 2-thioxo-6-aminothieno[3,2-d]pyrimidin-4-one derivatives could be easily accessible. Treatment of 49 with piperidine in the presence of the appropriate alkyl cyanide led to the aminothieno-fused derivatives 53a and 53b with good yields (Scheme 22) [39][30].
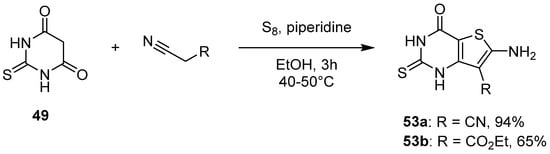
Scheme 22.
Synthesis of 2-thioxo-thieno[3,2-
d
]pyrimidines
53
by the Gewald reaction.
As shown in the previous examples, many access routes to these compounds are possible and allow to easily prepare a wide range of polysubstituted thienopyrimidines. Therefore, these compounds have been included in many biological studies. More particularly, their antiparasitic, antibacterial, antifungal and antiviral activities have been studied, and have been compiled in a entreviewy. For more details, see P. Lagardère et al. [131]
References
- Prisca Lagardère; Cyril Fersing; Nicolas Masurier; Vincent Lisowski; Thienopyrimidine: A Promising Scaffold to Access Anti-Infective Agents. Pharmaceuticals 2021, 15, 35, 10.3390/ph15010035.Pullarao, B.; Sharif, S.D.K.; Kumar, D.R.; Ramachandran, D. Design, Synthesis and Biological Evaluation of Thiophene Based Pyrimidin-4-One Derivatives as New Type of Antimicrobial Agents. Asian J. Chem. 2016, 28, 1997–2000.
- Ortikov, I.S.; Turdibaev, Z.É.; Islamova, Z.I.; Élmuradov, B.Z.; Abdurazakov, A.S.; Bektemirov, A.M.; Osipova, S.O.; Khushbaktova, Z.A.; Syrov, V.N.; Shakhidoyatov, K.M. Search for Bactericides Among Derivatives of Deoxyvasicinone, Mackinazolinone, and Thienopyrimidinones. Pharm. Chem. J. 2017, 51, 456–464.
- Habib, N.S.; Soliman, R.; El-Tombary, A.A.; El-Hawash, S.A.; Shaaban, O.G. Synthesis and Biological Evaluation of Novel Series of ThienoPyrimidine Derivatives as Anticancer and Antimicrobial Agents. Med. Chem. Res. 2013, 22, 3289–3308.
- Kanawade, S.B.; Toche, R.B.; Rajani, D.P. Synthetic Tactics of New Class of 4-AminothienoPyrimidine-6-Carbonitrile Derivatives Acting as Antimicrobial Agents. Eur. J. Med. Chem. 2013, 64, 314–320.
- Woodring, J.L.; Patel, G.; Erath, J.; Behera, R.; Lee, P.J.; Leed, S.E.; Rodriguez, A.; Sciotti, R.J.; Mensa-Wilmot, K.; Pollastri, M.P. Evaluation of Aromatic 6-Substituted Thienopyrimidines as Scaffolds against Parasites That Cause Trypanosomiasis, Leishmaniasis, and Malaria. Med. Chem. Commun. 2015, 6, 339–346.
- Shao, X.; AbdelKhalek, A.; Abutaleb, N.S.; Velagapudi, U.K.; Yoganathan, S.; Seleem, M.N.; Talele, T.T. Chemical Space Exploration around ThienoPyrimidin-4(3H)-One Scaffold Led to a Novel Class of Highly Active Clostridium Difficile Inhibitors. J. Med. Chem. 2019, 62, 9772–9791.
- Aly, H.M.; Saleh, N.M.; Elhady, H.A. Design and Synthesis of Some New Thiophene, Thienopyrimidine and Thienothiadiazine Derivatives of Antipyrine as Potential Antimicrobial Agents. Eur. J. Med. Chem. 2011, 46, 4566–4572.
- Desroches, J.; Kieffer, C.; Primas, N.; Hutter, S.; Gellis, A.; El-Kashef, H.; Rathelot, P.; Verhaeghe, P.; Azas, N.; Vanelle, P. Discovery of New Hit-Molecules Targeting Plasmodium Falciparum through a Global SAR Study of the 4-Substituted-2-Trichloromethylquinazoline Antiplasmodial Scaffold. Eur. J. Med. Chem. 2017, 125, 68–86.
- De Schutter, J.W.; Morrison, J.P.; Morrison, M.J.; Ciulli, A.; Imperiali, B. Targeting Bacillosamine Biosynthesis in Bacterial Pathogens: Development of Inhibitors to a Bacterial Amino-Sugar Acetyltransferase from Campylobacter Jejuni. J. Med. Chem. 2017, 60, 2099–2118.
- Mavrova, A.T.; Vuchev, D.; Anichina, K.; Vassilev, N. Synthesis, Antitrichinnellosis and Antiprotozoal Activity of Some Novel ThienoPyrimidin-4(3H)-Ones Containing Benzimidazole Ring. Eur. J. Med. Chem. 2010, 45, 5856–5861.
- Kim, J.; Kwon, J.; Lee, D.; Jo, S.; Park, D.-S.; Choi, J.; Park, E.; Hwang, J.Y.; Ko, Y.; Choi, I.; et al. Serendipitous Discovery of 2-((Phenylsulfonyl)Methyl)-ThienoPyrimidine Derivatives as Novel HIV-1 Replication Inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 5473–5477.
- Sławiński, J.; Żołnowska, B.; Pirska, D.; Kędzia, A.; Kwapisz, E. Synthesis and Antibacterial Activity of Novel 4-Chloro-2-Mercaptobenzenesulfonamide Derivatives. J. Enzym. Inhib. Med. Chem. 2013, 28, 41–51.
- Patel, A. Modi Synthesis and Biological Evaluation of Schiff Base Involving Thieno Pyrimidine Moiety as Antimicrobial Agents. RJLBPCS 2019, 5, 31–41.
- Giri, T.; Sailaja, G.; Laxminarayana, E.; Thirumala Chary, M.; Ramesh, M. Synthesis and Antibacterial Activity of Novel 4--Benzohydrazide Derivatives. Russ. J. Gen. Chem. 2017, 87, 1275–1280.
- Tharikoppula, G.; Eppakayala, L.; Maringanti, T.C.; Kamalapuram, C.; Kudle, K.R. Synthesis and Antibacterial Activity of Thienopyrimidine Amide Derivatives. Asian J. Chem. 2017, 29, 1515–1521.
- Temburnikar, K.W.; Zimmermann, S.C.; Kim, N.T.; Ross, C.R.; Gelbmann, C.; Salomon, C.E.; Wilson, G.M.; Balzarini, J.; Seley-Radtke, K.L. Antiproliferative Activities of Halogenated Thienopyrimidines. Bioorganic Med. Chem. 2014, 22, 2113–2122.
- Prabhakar, V.; Babu Kondra, S.; Maddula, S.R. Synthesis, Structural Elucidation of Novel ThienoPyrimidine Core Unit Containing 1,2,4-Triazoles and Thiophenes as Potent Antimicrobial Activity. Org. Chem. Curr. Res. 2016, 5, 1000169.
- Prabhakar, V.; Babu, S.J.; Siva Jyothi, S.V.L. Synthesis, Structural Elucidation and Anti-Bacterial Evaluation of Some Novel Heterocyclic Molecules Derived from Thienopyrimidine as a Core Unit. Org. Chem. Curr. Res. 2016, 5, 1000172.
- Sang, Y.; Han, S.; Han, S.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F. Follow On-Based Optimization of the Biphenyl-DAPYs as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors against the Wild-Type and Mutant Strains. Bioorganic Chem. 2019, 89, 102974.
- Al-Harbi, R.A.K.; Abdel-Rahman, A.A.H. Synthesis and Anti-HBV Activity of 2-(Methylthio)ThienoPyrimidin- 4(1H)-One Analogues of ACV. Der Pharma Chem. 2013, 5, 1–7.
- Kankanala, J.; Kirby, K.A.; Huber, A.D.; Casey, M.C.; Wilson, D.J.; Sarafianos, S.G.; Wang, Z. Design, Synthesis and Biological Evaluations of N-Hydroxy Thienopyrimidine-2,4-Diones as Inhibitors of HIV Reverse Transcriptase-Associated RNase H. Eur. J. Med. Chem. 2017, 141, 149–161.
- Abu-Hashem, A.A.; Abu-Zied, K.M.; El-Shehry, M.F. Synthetic Utility of Bifunctional Thiophene Derivatives and Antimicrobial Evaluation of the Newly Synthesized Agents. Mon. Chem. 2011, 142, 539–545.
- Dewal, M.B.; Wani, A.S.; Vidaillac, C.; Oupický, D.; Rybak, M.J.; Firestine, S.M. ThienoPyrimidinedione Derivatives as Antibacterial Agents. Eur. J. Med. Chem. 2012, 51, 145–153.
- Shaaban, O.G.; Issa, D.A.E.; El-Tombary, A.A.; Abd El Wahab, S.M.; Abdel Wahab, A.E.; Abdelwahab, I.A. Synthesis and Molecular Docking Study of Some 3,4-DihydrothienoPyrimidine Derivatives as Potential Antimicrobial Agents. Bioorganic Chem. 2019, 88, 102934.
- Endo, Y.; Kawai, K.; Asano, T.; Amano, S.; Asanuma, Y.; Sawada, K.; Onodera, Y.; Ueo, N.; Takahashi, N.; Sonoda, Y.; et al. 2-(Isopropylamino)ThienoPyrimidin-4(3H)-One Derivatives as Selective Phosphodiesterase 7 Inhibitors with Potent in Vivo Efficacy. Bioorganic Med. Chem. Lett. 2015, 25, 1910–1914.
- González Cabrera, D.; Le Manach, C.; Douelle, F.; Younis, Y.; Feng, T.-S.; Paquet, T.; Nchinda, A.T.; Street, L.J.; Taylor, D.; de Kock, C.; et al. 2,4-Diaminothienopyrimidines as Orally Active Antimalarial Agents. J. Med. Chem. 2014, 57, 1014–1022.
- Cohen, A.; Suzanne, P.; Lancelot, J.-C.; Verhaeghe, P.; Lesnard, A.; Basmaciyan, L.; Hutter, S.; Laget, M.; Dumètre, A.; Paloque, L.; et al. Discovery of New Thienopyrimidinone Derivatives Displaying Antimalarial Properties toward Both Erythrocytic and Hepatic Stages of Plasmodium. Eur. J. Med. Chem. 2015, 95, 16–28.
- Chambhare, R.V.; Khadse, B.G.; Bobde, A.S.; Bahekar, R.H. Synthesis and Preliminary Evaluation of Some N-Pyrimidin-3-Yl]-Carboxamide and 3-Substituted-5-(2-Furanyl)-2-Methyl-3H-ThienoPyrimidin-4-Ones as Antimicrobial Agents. Eur. J. Med. Chem. 2003, 38, 89–100.
- Abdel Hamid, A.M.; Shehta, W. Synthesis of Some Novel Furan-Tagged Thienopyrimidine Derivatives as Antibacterial Agents: Synthesis of Some Novel Furan-Tagged Thienopyrimidine Derivatives as Antibacterial Agents. J. Heterocycl. Chem. 2019, 56, 485–492.
- Aly, H.M.; Saleh, N.M. Utility of a Pyrimidine Thione Derivative in the Synthesis of New Fused PyrimidoPyrimidine, PyridoPyrimidine and Different Types of Thienopyrimidine Derivatives. Int. J. Adv. Res. 2014, 2, 694–702.
More