3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoAR) is an enzyme involved in the biosynthesis of cholesterol and an important target in the treatment of hypercholesterolemia.
- HMG-CoAR
- Cholesterol
- Hypercholesterolemia
1. Introduction
This entry introduces the enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoAR). This enzyme catalyzes one of the most important steps in cholesterol biosynthesis and is regarded as one of the most important drug targets in the treatment of hypercholesterolemia.
2. Biosynthesis of Cholesterol and the Role of HMG-CoA
In a detailed mechanism, the synthesis of cholesterol starts with the condensation of two acetyl-coenzyme A (acetyl-CoA) molecules to form the intermediate acetoacetyl-CoA. This process is catalyzed by a thiolase enzyme, known as acetyl-CoA acetyltransferase (ACAT). Hereinafter, the reaction of two acetoacetyl-CoA molecules catalyzed by HMG-CoA synthase (HMG-CoAS) allows for the formation of 3 hydroxy-3-methylglutaryl CoA (HMG-CoA), which is subsequently reduced to mevalonate by the enzyme HMG-CoA reductase (HMG-CoAR) and two NADPH molecules that function as cofactors (Figure 1). This latter reaction is the rate-limiting step of the overall synthesis of cholesterol, and it has been extensively studied since it defines the course of the reaction, known as the committed step, using HMG-CoAR as a regulatory enzyme.
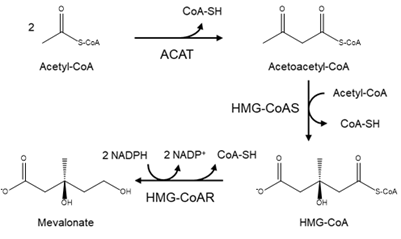
Figure 1. Diagram of the mevalonate pathway, the first stage in the biosynthesis of cholesterol.
The next step in the biosynthesis of cholesterol is the conversion of the intermediate mevalonate into two activated isoprenoids—isopentenyl-5-pyrophosphate and dimethylallyl pyrophosphate (IPP)—by the addition of three ATP molecules. The obtained isoprenoids are used to synthesize several biomolecules, such as cholesterol, ubiquinone and sterol-based hormones in animals; carotenoids in plants; and cell-wall components (such as undecaprenyl phosphate) in eubacteria [1][2][3][4][5][6][19,21–25].
A successive condensation reaction of activated isoprenes allows for the formation of a 30-carbon molecule, known as squalene, whose linear structure can be linked to the cyclic steroids and act as the biochemical precursor of all steroids. The synthesis of cholesterol is achieved by the mono oxidation of squalene into an epoxide using a NADPH molecule, followed by the cyclization of the intermediate to form a 4-ring compound, the lanosterol molecule, which is converted into cholesterol after multiple subsequent reactions.
Since cholesterol biosynthesis is an energetically complex and expensive process (involves 18 ATP molecules), it is only natural to assume that it is carefully regulated. In mammals, its regulation is controlled by the intracellular cholesterol concentration and the presence of the hormones insulin and glucagon [1][19]. As mentioned above, the most important checkpoint is the conversion of HMG-CoA to mevalonate, a step catalyzed by HMG-CoAR. The importance of this enzyme to the mevalonate pathway has been evaluated through various experimental works, such as the work of Chappell et al., in which, after introducing a constitutively expressed HMG-CoAR gene from a hamster into tobacco plants, the activity of the enzyme became unregulated and the accumulation of sterols increased 3–10-fold [7][26]. The studies surrounding this enzyme were performed to assess not only its importance in the biosynthesis of cholesterol and other steroids, but also to understand how it works and is regulated.
3. Structure of HMG-CoA Reductase
HMG-CoAR is a very important enzyme in the production and regulation of several essential compounds. As discussed above, mevalonate will lead to the formation of isoprenoids, which are a metabolite of major importance in several different organisms, ranging from bacteria to plants and animals [2][8][21,27]. For this reason, these organisms also have similar enzymes that perform analogous reactions. HMG-CoAR can be found in eukaryotes and some prokaryotes, with some differences observed. With a sequence analysis of this protein, it was possible to point out two different classes. Class I enzymes (EC: 1.1.1.34) are present in most archaea and eukaryotes and are inserted in the membrane of the endoplasmic reticulum (ER) [8][9][27,28]. Class II HMG-CoAR (EC: 1.1.1.88), on the other hand, is completely soluble in the cytoplasm and is found in some archaea and prokaryotes [10][29].
In a more detailed view, Class I enzymes have a transmembrane domain, ranging from residues 1 to 339; a cytosolic domain in which the active site is located (residues 460–888); and are connected by a linker region, which is comprised by residues 340–459 (the description of the residues is associated with human protein) [11][30]. Class II contains only the catalytic domain of the Class I enzyme, thus resulting in a 428-residue polypeptide [11][12][13][31–33]. This results in sequence identities ranging 14–20% [8][11][13][27,30,33]. Despite this low sequence homology between classes, as well as a different protein architecture, both enzymes have comparable positions for the active sites residues and a highly conserved catalytic portion (Figure 2) [11][13][30,33]. The study that led to the early development and understanding of the mevalonate pathway used this feature, as the first Class II HMG-CoAR enzyme was obtained from the eubacterium Pseudomonas mevalonii [14][15][34,35]. This enzyme was a useful model for the characterization of the active site residues, even preceding crystallographic information [14][15][16][17][18] [34–38].
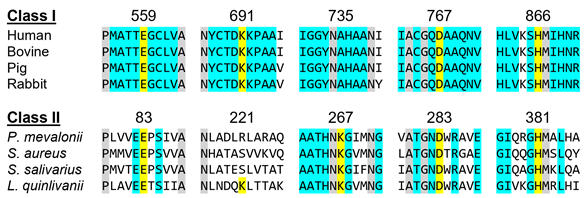
Figure 2. HMGCRs sequence alignment around the main catalytic residues, which are highlighted in yellow. The numbering is related to the human enzyme for Class I and with the Pseudomonas mevalonii one for Class II. Residues colored in grey are conserved between classes and those colored in light blue are conserved within the same class.
The tridimensional structural information for the human HMGCR (Figure 3) was solved for the first time in 2000 by Istvan et al. [18][38]. Currently, there are 22 structures for the catalytic domain of the human enzyme available on the Protein Data Bank, which sum up to more than 40 if we also count those from other organisms. These structures contain those of the enzyme alone as well as complexes between HMGCR and HMG-CoA, NADP+ and different statins .
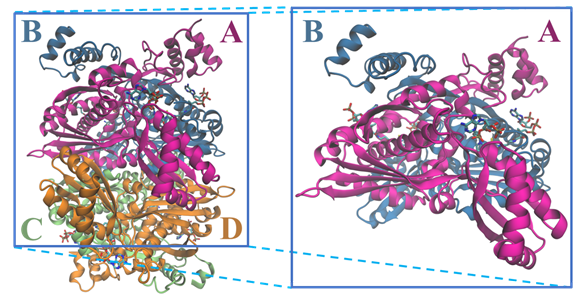
Figure 3. Structure of the tetramer and respective dimer of the human HMGCR, showing both CoA and NADPH (PDB entry 1DQA); chains are colored according with the letters: chain A is in magenta, chain B in blue, chain C in light green and chain D in orange.
3.1. Active Site Architecture and Catalytic Mechanism of HMG-CoA Reductase
The description of the active site of HMG-CoAR, enzyme responsible for the reduction of HMG-CoA to mevalonate, is briefly depicted above. In short, the active site of this enzyme is formed by two different subunits that form a dimer when bound together. The catalytic domain remains unchanged even with the formation of the tetramer.
The active site of the HMG-CoAR enzyme is a large cavity which is located at the monomer–monomer interface. This active site can be divided into three different binding subsites: one for HMG, one for CoA and one for NADPH.
The binding site of CoA (Figure 4b) is positioned in the L-domain of one monomer. The ADP moiety of CoA binds near the surface of the enzyme, in a pocket lined with positively charged residues. The residues that are involved in the CoA binding pocket are Ser565, Asn567, Arg568, Lys722, Ser865, His866 and Tyr479 (this last residue comes from the neighboring monomer). The side chain of Tyr479 is of particular importance, as it interacts with the adenine base of CoA through π–π stacking while its hydroxyl group makes a hydrogen bond with the 3’-phosphate of the ribose moiety [19] [49].
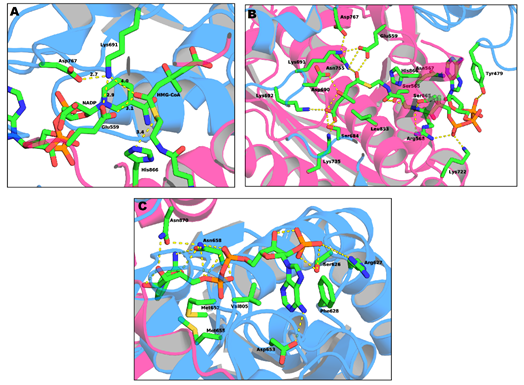
Figure 4. Representation of: (a) the active site of HMG-CoAR; (b) the binding sites of the HMG portion of HMG-CoA; and (c) the binding sites of NADP (PDB entry 1DQ9).
The NADPH (Figure 4c) binds to the S-domain of the opposing subunit in which the HMG-CoA binding pocket is located. Residues Ser626, Arg627, Phe628, Asp653, Met655, Gly656, Met657, Asn658 and Val805 come from the S-domain and residues Asn870 and Arg871 from the neighboring monomer. In the presence of NADPH, a conformational change in the C-terminus of the enzyme is observed which leads to the closure of the active site [19][49].
Lastly, the HMG binding site (Figure 4a) is located at the interface of the two monomers. This binding pocket is formed by residues Ser684, Asp690, Lys691, Lys692 and Asp767 from one subunit and Glu559, Lys735, Asn755, Leu853 and His866 from the other. This is also the catalytic site of the enzyme. The currently accepted mechanism of HMG-CoA is present in Figure 5.
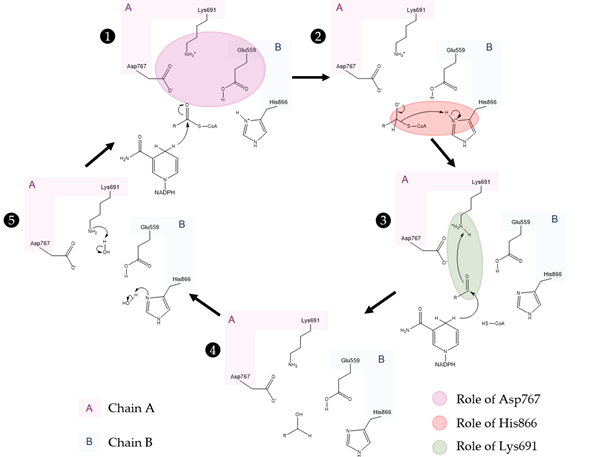
Figure 5. Currently accepted catalytic mechanism of HMG-CoAR [56]. The catalytic residues are Lys691, Glu559, Asp767 and His866; chain A is highlighted in pink and chain B is highlighted in blue. Glu559 and His866 are restored in a following step by deprotonation of adjacent water molecules.
As observed in the mechanism shown in Figure 5, the enzyme HMG-CoAR enables the reduction of thioesterified HMG-CoA to mevalonate, using two molecules of NADPH for successive hydride transfers. The first step results in the formation of mevaldyl-CoA hemi-thioacetal, followed by the formation of the mevaldehyde, which occurs with the collapse of the thiohemiacetal and formation of CoA-SH (protonation of the thiol anion by Hys866). In the final step, the second NADPH reduces mevaldehyde to mevalonate [8][13][19][20][21][22][20,27,33,48,49,56].
The perception of this complex mechanism has changed through the years. In an earlier work, Tabernero et al. proposed a similar mechanism to the one depicted in Figure 5 for the P. mevalonii enzyme, in which Lys267 was proposed to be the general acid that stabilizes the mevaldyl-CoA intermediate [21][48]. As shown in Figure 3 and their work, it is not obvious to find the correct association between this residue from P. mevalonii and other HMG-CoARs using sequence homology. This was also observed by Haines et al., as their alignment is different from ours and the previous work [8][27]. In this latter work, they observed that the reduction of mevaldyl-CoA was performed with the aid of Glu83 (which is equivalent to Glu559 in the human enzyme), instead of Lys267. This work is supported by the position of the active site human residues as: (i) negatively charged intermediates can be stabilized by the positive charge of Lys691; (ii) the proximity between one of the side chain oxygens of Glu559 and the carbonyl oxygen of HMG suggests that this residue is protonated; (iii) the negatively charged Asp767 plays a critical role, as it is close enough to the glutamate to influence its pKa value, as well as being able to stabilize through ionic interactions the Lys691 side chain in the active; and (iv) the His866 can make a hydrogen bond with CoA thiol [8][27]. The mechanism proposed in Figure 5 follows the line of an QM/MM study, in which several approaches were tested. Oliveira et al. concluded that the correct mechanism is similar to the one proposed by Tabernero, however some structural features of Haines were used [22][56]. The presence of a neutral Glu559 in the active site allows for this residue to be more distant from the positively charged His866, as Glu559 and Asp767 form a hydrogen bond. Thus, Glu599 is no longer in an appropriate position to stabilize the mevaldyl-CoA intermediate, as previously suggested [8][27]. From the models tested by Oliveira et al., the one with the neutral Glu599 presented the lowest activation free energy for the reaction mechanism in Figure 5, in which the protonation of mevaldyl-CoA is performed by Lys691 [22][56].
Considering the structural differences between both classes of HMG-CoAR, and, consequently the different positions of the catalytic residues, it is safe to assume that the reaction mechanisms will be different. P. mevalonii enzyme is responsible for the reverse reaction of the human variant, and it has been shown that it is possible for this prokaryote to grow on mevalonate only [23][57]. The cis-loop found in Class I reductases, which is key for the positioning of the catalytic residues, is missing in this version of the protein. Instead, the position of His381 (analogous to His866) is approximated to the active site by the closure of the flap domain, i.e., the C-terminal 50 residues of Class II. This motion also alters the binding of the substrate and the cofactor [19][24][49,58].