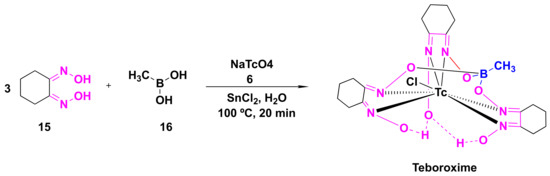
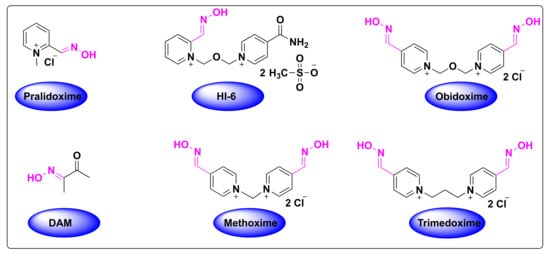
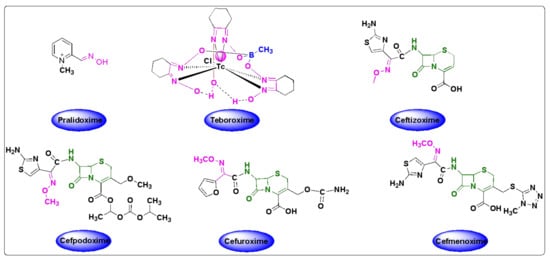
Organophosphate (OP) poisoning continues to be a major threat to humans. Oximes represent the most important class in medicinal chemistry, renowned for their widespread applications as OP antidotes, drugs and intermediates for the synthesis of several pharmacological derivatives. Common oxime based reactivators or nerve antidotes include pralidoxime, obidoxime, HI-6, trimedoxime and methoxime, among which pralidoxime is the only FDA-approved drug. Cephalosporins are β-lactam based antibiotics and serve as widely acclaimed tools in fighting bacterial infections. Oxime based cephalosporins have emerged as an important class of drugs with improved efficacy and a broad spectrum of anti-microbial activity against Gram-positive and Gram-negative pathogens.
- oximes
- organophosphates
- acetylcholinesterase
- antidotes
- pralidoxime
- obidoxime
- HI-6
- trimedoxime
- methoxime
- cefuroxime
- ceftizoxime
- cefpodoxime
- cefmenoxime
- antibiotics
- cephalosporins
1. Introduction
1.1. The Sources and Discovery of Oximes
1.2. The Synthesis of Oximes
Oximes have gained wide interest in the field of organic chemistry due to their ease of synthesis from the carbonyl compounds, and their facile conversion to nitriles, amines, nitro compounds and other heterocyclic compounds [13][18]. In addition, they are popular as protecting groups of carbonyl compounds and as intermediates in the Beckmann rearrangement for the synthesis of important β-lactam derivatives [14][19]. Oximes have wide ranging industrial applications and a classic example of their significance is indicated by the great production of caprolactam, which is a precursor to nylon-6, whose production exceeds over five million tons per year [15][20].1.3. Isomerism of Oximes
Oximes generally exist in the form of interconvertible E and Z stereoisomers, depending on the orientation of groups around the C=N bond [16][24]. These isomers can have different physical properties and can be readily isolated by chromatography or recrystallization techniques. Oxime stereoisomers have important pharmacological properties and studies revealed that Z-isomers are more stable and predominant than E-isomers. Differential scanning calorimetry studies also proved this fact that heating and melting of E-oxime readily afforded the Z-oxime. Interestingly, the reagents used for the synthesis of oximes from aldehydes and ketones can also catalyze the interconversion of E and Z isomers. Temperature plays a critical role in the determination of the isomer ratio, as any change in the temperature can vary the position of equilibrium and subsequently alter the equilibration ratio of the isomer mixture [17][25].2. Classification of FDA-Approved Oximes
Despite the growing number of oxime-based compounds that have passed clinical testing and are already widely used for medicinal purposes, the FDA approved only six of them, which include pralidoxime, teboroxime (a myocardial imaging agent) and four oxime-based cephalosporines.2.1. Oximes as Organophosphate (OP) Poisoning Antidotes
Tetraethylpyrophosphate (TEPP) was the first OP, synthesized in 1854 by the French chemist Philippe de Clermont [18][35]. The general structure of OPs was first provided by Schrader in 1937, which is comprised of esters or thioesters or anhydride derivatives of phosphoric acid [19][36]. OPs contain a facile leaving group, which is prone to hydrolysis and is eliminated by the phosphorylation of acetylcholinesterase [20][37].2.2. Significance of Acetylcholinesterase and Its Inhibition in OP Poisoning
Acetylcholinesterase (AChE, also known as acetylcholinehydrolase) is an enzyme that catalyzes the hydrolysis of neurotransmitter acetylcholine to choline and acetic acid (or acetate) [21][53]. Acetylcholine is a major neurotransmitter found in the central and peripheral nervous systems, best known for the transmission at the neuromuscular junction and at ganglionic synapses, and is broken down by the action of AChE [19][36]. AChE is composed of a serine site and an anionic site. The serine site is embedded in the active site of the enzyme and is prone to OP attack, ultimately resulting in the phosphorylation of serine site and the inactivation of AChE through the formation of a strong covalent bond. This irreversible binding results in the AChE inhibition by excessive cholinergic stimulation on the nicotinic and muscarinic receptors. This overstimulation can result in a variety of symptoms based on the type of receptor. Hyperstimulation of muscarinic receptors can lead to nausea, vomiting, diarrhea, seizures, abdominal cramps, incontinence of feces and urine, while the hyperactivation of nicotinic receptors can result in hypertension, muscle cramps, fasciculations, tachycardia and paralysis [22][54]. Death in patients due to OP poisoning, commonly occurs from the respiratory failure.2.3. Detoxification of OP Poisoning by Pralidoxime
The inactivation of AChE by OPs leads to the accumulation of acetylcholine in the exposed patients, resulting in the overactivation of muscarinic and nicotinic receptors, which ultimately culminates in the range of aforementioned symptoms [23][55]. The mechanism of action of the reactivation of AChE by oximes can be best described using the FDA-approved drug, 2-pyridine-aldoxime methyl chloride (2-PAM) (also known as pralidoxime), as a classic example (Figure 37). The 2-PAM is the most popular and well-investigated agent, along with HI-6 and obidoxime [4][19][24][4,36,56]. The 2-PAM has a positively charged quaternary nitrogen atom of the pyridinium ion that specifically interacts with the anionic site of the AChE, and, then, the hydroxyl group of the oxime moiety replaces the serine residue by nucleophilic substitution, releasing the free hydroxy (OH)-group of the amino acid (Figure 37) [25][26][27][57,58,59]. This can be explained due to the higher affinity of 2-PAM to become phosphorylated over the serine residues by the OP agent, and thereby liberating the enzyme and making it available to bind to acetylcholine [21][53]. This AChE restoration relieves the associated symptoms of OP poisoning, such as muscle twitching and tremors, convulsions, difficulty breathing and fatigue [19][36].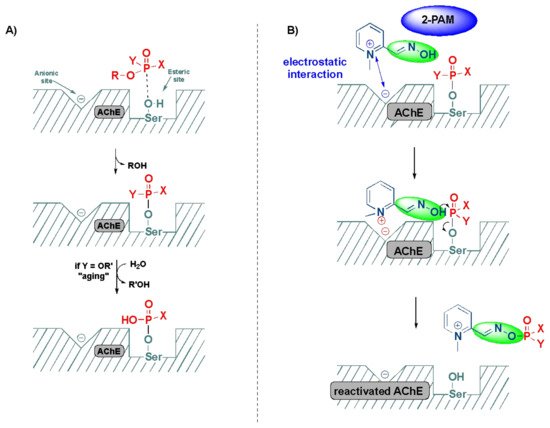
34. Oxime Based FDA Approved Drugs
3.1. Pralidoxime
4.1. Pralidoxime
3.2. Teboroxime
4.2. Teboroxime
Teboroxime or technetium-99m-teboroxime (Tc-99m-teboroxime) is a small, neutral, lipophilic mixture obtained from the boronic acid adducts of technetium dioxime complexes (BATOs) [30][31][32][94,95,96]. It is synthesized from methylboronic acid, stannous chloride and 99m-technetium salts following the synthetic route, as shown in Scheme 12. It is used as a myocardial perfusion imaging agent, ever since FDA approval in the year 1991 [33][34][97,98]. Tc-99m-teboroxime was then withdrawn from the market because of rapid clearance kinetics that did not allow high qualities of the images [35][85]. Nevertheless, it was taken back to the market, in some countries, for the solid-state photon emission computed tomography (SPECT). Its pharmacokinetics and distribution were discussed in relation to coronary artery disease and ischemia [36][37][99,100].