Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Elisabetta Rovida and Version 2 by Catherine Yang.
Extracellular signal-regulated kinase 5 (ERK5) belongs to the mitogen-activated protein kinase (MAPK) family that consists of highly conserved enzymes expressed in all eukaryotic cells and elicits several biological responses, including cell survival, proliferation, migration, and differentiation.
- ERK5
- cancer
- cell proliferation
- targeted therapy
- resistance mechanism
- BMK1
- MAPK7
1. The ERK5 Signaling Pathway
ERK5 (also referred to as big mitogen-activated protein kinase 1, BMK-1) is encoded by the MAPK7 gene and is a member of the MAPK family. ERK5 is ubiquitously expressed in mammalian tissues and cell types, where it is activated by extracellular stimuli, including several growth factors and cellular stresses [1][2][3][4][2,3,4,5]. Human ERK5 protein contains 816 amino acids and consists of an N-terminal kinase domain (78–406 aa) and a unique C-terminal tail (410–816 aa), which harbors an autoinhibitory function [5][6]. The C-terminus also contains a myocyte enhancer factor 2 (MEF-2)-interacting region (440–501 aa) [6][7], a nuclear localization signal (NLS) (505–539 aa), and a transcriptional activation domain (664–789 aa) [6][7], which associate with and activate several transcription factors [7][8]. Activation of ERK5 requires dual phosphorylation of threonine and tyrosine residues within a TEY motif in the activation loop of the kinase domain [8][9]. At this site, ERK5 can be phosphorylated and activated by MEK5, which has a unique specificity for ERK5. Activation by MEK5 induces an open conformation of ERK5, the exposure of the NLS, and the translocation into the nucleus. The latter event is crucial for the proliferative signals induced by ERK5 [9][10]. Besides being phosphorylated at the TEY motif, ERK5 is able to phosphorylate its C-terminal tail on serine and threonine residues. These residues at the C-terminus have also been reported to be phosphorylated by CDK1 and/or ERK1/2 [10][11]. Upstream activators of MEK5–ERK5 are MEKK2 and MEKK3, as well as SRC [11][12], TPL2/COT, RAS, and AKT [12][13]. Known substrates for ERK5 are transcription factors, including c-FOS, c-MYC, Sap-1a and MEF2A, C and D, and other kinases, such as RSK and serum/glucocorticoid-regulated kinase (SGK) (Figure 1) [13][14].
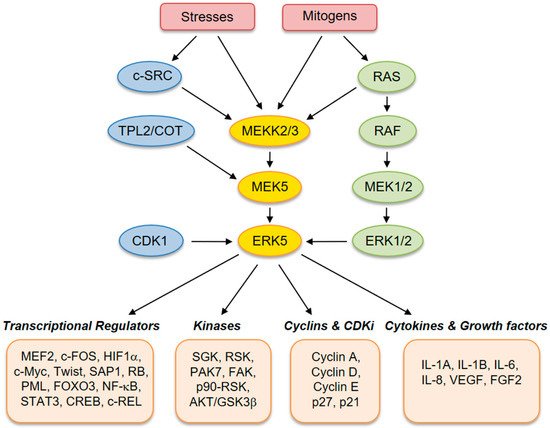
Figure 1. Schematic representation of the MEK5–extracellular signal-regulated kinase 5 (ERK5) pathway with activators and downstream effectors.
2. Sustaining Proliferative Signals
ERK5 plays a well-established role in cell proliferation. Several reports have shown activation of ERK5 in response to several mitogens, including epidermal growth factor (EGF) [14][15], nerve growth factor [15][16], fibroblast growth factor (FGF) [16][17], colony-stimulating factor-1 [17][18], and platelet-derived growth factor (PDGF) [18][19]. ERK5 regulates different phases of the cell cycle. For instance, ERK5 mediates G1/S transition by regulating the expression of cyclin D1. Conversely, ERK5 inhibition decreases serum-induced cyclin D1 expression [19][20]. Furthermore, ERK5 is implicated in G2/M transition and is required for mitotic entry. The induction of G2/M by ERK5 depends on the activation of the transcription factor NF-kB, which in turn upregulates mitosis-promoting genes, such as cyclins B1 and B2 and CDC25B [20][21][21,22].
During the last few years, several studies have demonstrated the critical role of MEK5–ERK5 signaling in cancer cell proliferation and tumorigenesis (Figure 2). The role of ERK5 in prostate cancer (PC) proliferation is well established. Human PC displays aberrant expression of ERK5, with significant upregulation of ERK5 protein in high-grade tumors [22][23]. Increased ERK5 cytoplasmic positivity correlates with Gleason score, bone metastases, and locally advanced disease at diagnosis. Pointing to an important role of nuclear ERK5 in cancer, a subgroup of PC patients shows ERK5 nuclear localization, which correlates with poor disease survival [23][24]. Functionally, expression of a constitutively active form of MEK5 increases the percentage in the S phase of human PC LNCaP cells, leading to enhanced proliferation in vitro [22][23]. Along this line, overexpression of ERK5 in PC3 cells increases proliferation in vitro and xenograft growth in vivo [23][24], whereas ERK5 silencing suppresses PC3 cell proliferation [24][25]. In addition, EGF-mediated ERK5 activation induces proliferation of RWPE-2 and PC3 cells by promoting entry into the S phase through upregulation of cyclins A and E [25][26]. Recently, phthalates have been shown to promote PC3 and 22RV1 PC cell proliferation through activation of ERK5 and p38, linking environmental pollution with ERK5 and cancer [26][27]. The role of microRNA as negative regulators of ERK5 is well documented and implicated in mediating ERK5-dependent PC cell proliferation. MiR-143 inversely correlates with nuclear ERK5 in human PC [27][28] and interferes with ERK5 signaling to abrogate PC progression in mice [28][29]. Similarly, overexpression of miR-143 suppresses proliferation of human bladder cancer T24 and Hela cells in vitro and reduces tumor growth of breast cancer (BC) cells in vivo through downregulation of ERK5 [29][30][31][30,31,32].
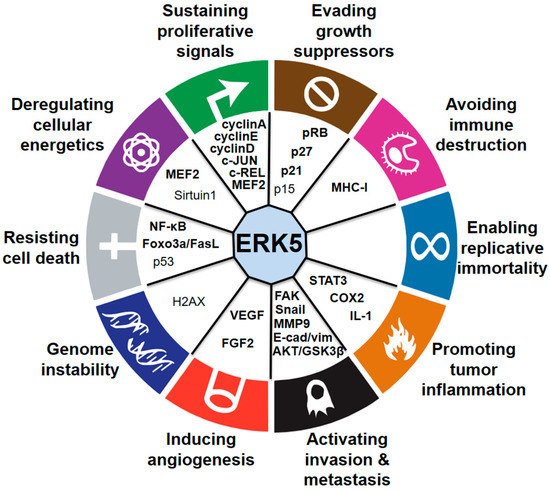
Figure 2. Connections between ERK5 and the hallmarks of cancer. Established (bold) and proposed (nonbold) downstream mediators of ERK5 are indicated.
3. Evading Growth Suppressors
Regarding the regulation of p21 by ERK5, it has been shown that activated ERK5 inhibits PML-dependent induction of p21. Using XMD8-92, it was later shown that ERK5 inhibition blocks tumor cell proliferation in vitro and significantly inhibits tumor growth in vivo, demonstrating the efficacy and tolerability of ERK5 targeting for cancer treatment in preclinical models [32][68]. Later on, the same group showed that ERK5-mediated phosphorylation of the tumor suppressor PML induces dissociation by MDM2, downregulating the expression of the p53 tumor suppressor [33][69]. Moreover, based on evidence obtained in MDA-MB-231 breast cancer cells, it was proposed that ERK5 blocks p21 expression via a mechanism that implicates c-MYC-dependent transcriptional regulation of the miR-17-92 cluster [34][66]. Regarding p27, we provided evidence that increased expression of p27 upon ERK5 inhibition is likely mediated by AKT/FOXO4. Indeed, XMD8-92 treatment resulted in the partial block of G1/S transition, decreased phosphorylation of AKT, and increased the level of p27 and nuclear FOXO4 [35][62]. Besides regulating p21 expression, inhibition of either MEK5 or ERK5 reduces RB phosphorylation in melanoma cells [36][63].
Interestingly, a link between ERK5 and the tumor suppressor ataxia telangiectasia mutated (ATM) has been identified. Indeed, in the absence of ERK5, the development of T-cell lymphoma in Atm−/− mice is reduced, revealing a functional interaction between ATM and ERK5 (see also the section “Genome Instability”) [37][70]. Transforming growth factor β (TGF-β) signaling is considered a suppressor of tumor growth at the initial stages of tumorigenesis. Despite ERK5 is a well-established mediator of TGF-β-elicited epithelial-to-mesenchymal transition (EMT), a clear tumor-promoting feature (see the section “Activating Invasion and Metastasis”), there is no evidence of a possible role of ERK5 in the regulation of TGF-β-elicited growth-suppressive effects.
4. Avoiding Immune Destruction
In recent years, several reports have highlighted a link between ERK5 signaling and the ability of tumors to evade the immune system. For instance, in human and mouse leukemic cells, ERK5 silencing downregulates major histocompatibility complex class I (MHC-I) expression at the plasma membrane. Injection of ERK5-silenced tumor cells in syngeneic mice recruits and activates natural killer (NK) cells and induces production of cytokines such as IFNγ and TNFα, which attract new NK cells to the site. This raises an immune response against wild-type tumor cells that attenuates tumorigenesis in vivo [38][34]. Similarly, restoration of miR-17/20a functions in murine breast cancer and colon cancer cells mediates the inhibition of the MEKK2–MEK5–ERK5 pathway, leading to the downregulation of MHC-I (H-2D) molecules expressed on tumor cells and enhancing the antitumor activity of NK cells in vivo [39][71].
Deletion of ERK5 in an established PTEN-deficient mouse model of PC increases T-cell infiltration [40][72]. A transcriptomic analysis revealed upregulation in the prostate of double-knockout mice of the chemokines CCL5 and CXCL10, two potent chemoattractants for T lymphocytes. Consistent with this effect, prostate epithelial and stroma of tumors from double-knockout mice show an increase of a predominantly CD4+ T-cell infiltrate. These data provide a preclinical proof of concept that targeting ERK5 might enhance T-cell infiltrates in PC, with possible implications for exploiting immunotherapy in this cancer type [40][72].
5. Enabling Replicative Immortality
The involvement of ERK5 in replicative immortality in cancer cells has not been reported. However, prolonged treatment of the neuroepithelial neuroblastoma/Ewing sarcoma SK-N-MC cell line with the telomerase inhibitor GRN163 upregulates the expression of miR-143 [41][73], a well-known negative regulator of ERK5 [28][29]. Based on that, we might speculate that ERK5 expression is affected by telomere shortening. On the other hand, cellular senescence, characterized by an irreversible arrest in cell proliferation, is among the biological processes that may prevent cellular immortality [42][74]. No definitive link has been demonstrated showing the role of ERK5 in this biological process. However, microarray data identified ERK5 among the genes responsible for the maintenance, but not onset, of methotrexate-induced cellular senescence in the human colon cancer C85 cell line [43][75].
6. Promoting Tumor Inflammation
ERK5 has been proposed as a mediator of cancer-associated inflammation in epidermal carcinogenesis. In particular, epidermal expression of ERK5 is required to mediate inflammation in the skin [44][76]. Indeed, inactivation of ERK5 in epidermal keratinocytes has been shown to prevent inflammation-driven tumorigenesis, with evidence that ERK5 is critical for the development of skin SCC, one of the most common types of human nonmelanoma skin cancer. The causal relationship between ERK5 and inflammation is further supported by the demonstration that epidermal ERK5 controls the expression of a specific subset of proinflammatory cytokines, including IL-1A, IL-1B, and COX-2. Importantly, suppression of inflammation by ERK5 deletion in neoplastic keratinocytes reduces tumor burden [44][76]. This study demonstrated for the first time that ERK5 in epithelial cells is able to remodel the inflammatory microenvironment to support cancer growth. Consistently, it has recently been shown that the ERK5 inhibitor XMD8-92 reduces mesothelioma tumor growth in vivo and inhibits the expression of inflammasome-related genes, such as Caspase-1, IL-1A, IL-1B, HMGB1, and PYCARD [45][77], although the mechanism of this regulation remains to be determined.
ERK5 has been shown to positively regulate, through its target MEF2C, factors important for monocytic differentiation in human myeloid leukemia cells [46][78]. Recently, Tournier’s group demonstrated that selective ERK5 ablation in macrophages blocks phosphorylation of signal transducer and activator of transcription 3 (STAT3), a transcription factor crucial for macrophage polarity, impairing the growth of melanoma and carcinoma xenografts. Furthermore, targeting ERK5 in macrophages induced a transcriptional switch in favor of proinflammatory mediators. This study suggests that blocking ERK5 may represent a treatment strategy to reprogram macrophages toward an antitumor state by inhibiting STAT3-induced gene expression [47][79]. Important clinical implications have emerged from this study. First, the possibility that targeting protumor macrophages via anti-ERK5 therapy appears to be an attractive strategy for cancer treatment. Second, there is the occurrence of a possible synergistic effect between targeted inhibition of ERK5 in macrophages with immune checkpoint inhibitors. This effect is in line with the notion that, in addition to contributing to tumorigenesis, TAM play a critical role in tumor-drug resistance and disease relapse after therapy [48][80].