Atherosclerotic cardiovascular diseases (ASCVD), including coronary artery disease, cerebrovascular disease, and peripheral arterial disease, represent a significant cause of premature death worldwide. Biomarkers, the evaluation of which would allow the detection of ASCVD at the earliest stage of development, are intensively sought. Moreover, from a clinical point of view, a valuable biomarker should also enable the assessment of the patient’s prognosis. It has been known for many years that the concentration of fibrinogen in plasma increases, inter alia, in patients with ASCVD. On the one hand, an increased plasma fibrinogen concentration may be the cause of the development of atherosclerotic lesions (increased risk of atherothrombosis); on the other hand, it may be a biomarker of ASCVD, as it is an acute phase protein. In addition, a number of genetic polymorphisms and post-translational modifications of fibrinogen were demonstrated that may contribute to the risk of ASCVD.
1. Fibrinogen—Physiological and Pathophysiological Aspects
Fibrinogen, coagulation factor I, is a 340 kDa glycoprotein that plays an important role in many physiological and biochemical processes. The name “fibrinogen” was used for the first time in 1847 by Rudolf Virchow, while in 1872, Alexander Schmidt indicated that the conversion of fibrinogen to fibrin is an enzymatic process
[1][9]. Physiologically, almost all fibrinogen is found in plasma, and its concentration is 1.5–3.5 g/L (normal concentration ranges may vary slightly among different laboratories), with a half-life (T
1/2) of 3–5 days
[2][3][10,11]. The plasma concentration of fibrinogen largely depends on the factors regulating its synthesis and genetic predisposition
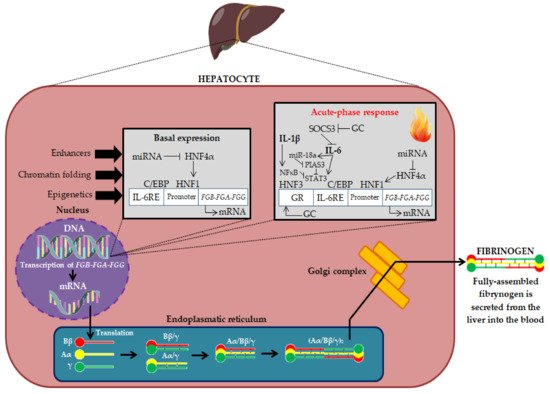
[2][3][10,11]. Fibrinogen is expressed primarily in hepatocytes and is regulated transcriptionally and post-transcriptionally. The biosynthesis of fibrinogen in the liver is primarily constitutive. Fibrinogen synthesis is regulated by acute-phase proteins, mainly by IL-6 (derived by monocytes, macrophages, and vascular endothelial cells), which induces its synthesis in the liver, while IL-1β and tumor necrosis factor-alpha (TNF-α) suppress its synthesis
[2][4][10,12]. Thus, fibrinogen is an acute phase protein because its biosynthesis is increased during inflammation (in the acute phase of inflammation, plasma fibrinogen concentrations can exceed 7 g/L). The production of fibrinogen is also increased by glucocorticosteroids (GC) (
Figure 1)
[2][10].
Figure 1. Fibrinogen biosynthesis in liver hepatocytes
[1][2][3][9,10,11]. DNA—deoxyribonucleic acid; mRNA—Messenger ribonucleic acid; FGA-FGB-FGG—Fibrinogen chain genes; miRNA—Micro RNA; HNF4α—Hepatocyte Nuclear Factor 4 Alpha; HNF1—Hepatocyte nuclear factor 1; C/EBP—CCAAT enhancer binding proteins; IL-6RE—IL-6 response element; SOCS3—Suppressor of cytokine signaling 3; miR-18a—microRNA-18a; NFκB—Nuclear factor kappa-light-chain-enhancer of activated B cells; PIAS3—Protein inhibitor of activated STAT 3; STAT3—Signal transducer and activator of transcription 3; HNF3—Hepatocyte nuclear factor 3; IL-1β—Interleukin 1β, IL-6—Interleukin 6; GR—Glucocorticoid receptor; GC—Glucocorticosteroids; Bβ, Aα and γ—Fibrinogen polypeptide chains.
Fibrinogen biosynthesis begins with the expression of the three genes
FGA,
FGB and
FGG, clustered in a 50 kb region of human chromosome 4 (long arm; 4q31.3–4q32.1)
[2][10]. The mechanisms regulating the expression of fibrinogen genes are not fully understood. Genome-wide association studies (GWAS) identified single-nucleotide polymorphisms (SNP) within fibrinogen genes, as well as loci distinct from fibrinogen that implicate transcription factors (e.g., hepatocyte nuclear factors 1 and 4 (HNF1 and HNF4), signal transducer and activator of transcription 3 (STAT3) and inflammatory signaling pathways downstream of IL-6 in fibrinogen expression. In addition, microRNA (miR) from the hsa-miR-29 and hsa-miR-409-3p family reduce fibrinogen expression in hepatoma cells in vitro, revealing mechanisms that can fine-tune fibrinogen levels in response to environmental signals
[5][13]. The transcription of mRNA leads to the generation of three homologous polypeptide chains: Bβ, Aα and γ. The assembly of the six chains takes place in a stepwise manner, in which single chains assemble first into Aα/γ and Bβ/γ complexes, then into Aα/Bβ/γ half-molecules, and finally into hexameric complexes (Aα/Bβ/γ)
2 linked by disulfide bonds (
Figure 1)
[3][11]. Fibrinogen has a complex trinodular structure with a central nodule (E-domain) that contains the N-terminus of each chain and two lateral globular domains (D-domains) that contain the C-terminus of Bβ- and γ-chains. The E-domain is linked to the two D-domains by a coiled-coil triple helix structure
[6][14].
Post-translational modifications of fibrinogen significantly affect its biological function. A systematic review by de Vries et al. summarized the knowledge of post-translational modifications of fibrinogen. A number of post-translational modifications were identified: oxidation, nitration, glycosylation, glycation, acetylation, phosphorylation, homocysteinylation, citrullinization, carbamylation and guanidinylation. These modifications were found to affect the rate of fibrin polymerization, the blood clot structure, and the course of fibrinolysis. Thus, post-translational modifications of fibrinogen may play an important role in the physiology and pathophysiology of blood coagulation
[7][15].
Moreover, genetic polymorphisms, which may influence the risk of various diseases, play an important role in the properties of fibrinogen
[8][16].
Many factors and/or conditions were shown to increase the plasma concentration of fibrinogen. These include: female gender, Black ethnicity, age, diabetes, smoking and alcohol consumption, arterial hypertension, obesity, lipid disorders, metabolic syndrome, menopause, oral contraceptives, microalbuminuria, lower socioeconomic status and premature family history of CVD. The association with body mass index (BMI) was twice as strong in women as in men. However, the association with smoking cigarettes was much stronger in men. Interestingly, plasma fibrinogen concentration is inversely related to serum HDL cholesterol (high-density lipoprotein cholesterol) concentration
[9][10][11][17,18,19]. Interestingly, the renin-angiotensin-aldosterone system (RAAS) plays an important role in the regulation of fibrinogen plasma concentration. Therefore, as pointed out by Kryczka et al., differences in the regulation of RAAS in women (including the effect of estrogens) and in men may affect the fibrinogen plasma concentration and observed clinical effect
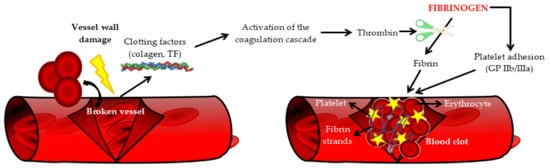
[12][20]. The basic physiological function of fibrinogen is participation in the final stage of the clotting process and transformation into a fibrillar protein—fibrin, which forms a blood clot (
Figure 2)
[3][11].
Figure 2. The role of fibrinogen in blood coagulation [13]. TF—Tissue factor, GP IIb/IIIa—Integrin receptors. The role of fibrinogen in blood coagulation [21]. TF—Tissue factor, GP IIb/IIIa—Integrin receptors.
Moreover, fibrinogen is involved in the matrix physiology (by interaction with plasminogen, FXIII, vitronectin and fibronectin), regulation of the inflammatory process, infection, wound healing, intercellular interaction, cell migration, tumor growth, angiogenesis, and metastasis
[3][11].
The increased fibrinogen plasma concentration directly activates many mechanisms, which, consequently, may intensify the progression of atherosclerosis
[12][20]. The pro-atherogenic mechanisms of action of fibrinogen are summarized in
Table 1.
Table 1. The pro-atherogenic mechanisms of action of fibrinogen
[12][20]. IL-1—Interleukin 1; TNF-α—Tumor necrosis factor α; IL-8—Interleukin 8; MCP-1—Monocyte chemoattractant protein-1; GP IIb/IIIa—Integrin receptors; IL-1β—Interleukin aβ; ICAM-1—Intercellular adhesion molecule 1; EC—Endothelial cells; LDL—Low-density lipoprotein; SMC—Smooth muscle cell.
| Main Pro-Atherogenic Properties of Fibrinogen |
|
|
- ✓
-
↑ Severity of inflammation: promotes an inflammatory response by inducing the exposition of proinflammatory cytokines on monocytes (IL-1 and TNF-α) as well as chemokines, such as IL-8 and MCP-1, on endothelium and fibroblasts, which promote atherosclerotic plaque formation;
- ✓
-
Activation of platelets (via GP IIb/IIIa receptors) leading to the production of the pro-inflammatory cytokines, CD40 ligand and IL-1β, which promote atherosclerotic plaque formation;
- ✓
-
↑ Expression of adhesion molecules (ICAM-1) on vascular EC leading to the adhesion of leukocytes, macrophages and platelets;
- ✓
-
↑ Production of vasoactive factors by EC leading to an increase in its permeability and impairing its vasorelaxant properties;
- ✓
-
Accumulation of fibrinogen in the vessel wall enhances the infiltration of macrophages, which are precursors of foam cells;
- ✓
-
The accumulated fibrinogen deposits in the vessel wall absorb LDL cholesterol, which leads to the formation and growth of atherosclerotic plaque;
- ✓
-
Increasing the adhesion of neutrophils to activated platelets attached to the injured arterial wall, which promotes the formation of atherosclerotic plaque;
- ✓
-
↑ SMC migration and proliferation, as well as stimulation of angiogenesis.
|
|
In summary, fibrinogen has many important functions in human physiology, but also many unfavorable pathophysiological pathways induced by increased fibrinogen plasma concentration were described, which aggravate the atherosclerotic process.
2. Fibrinogen Molecular Modifications and Cardiovascular Risk
A number of molecular modifications, such as gene polymorphisms, alternative splicing and post-translational modifications, may influence the plasma concentration of fibrinogen and its biochemical properties, and consequently the risk of CVD. As mentioned earlier, fibrinogen biosynthesis begins with the expression of the three genes
FGA,
FGB and
FGG. Many studies indicate the significant role of modifications at the stage of fibrinogen biosynthesis in shaping the risk of CVD.
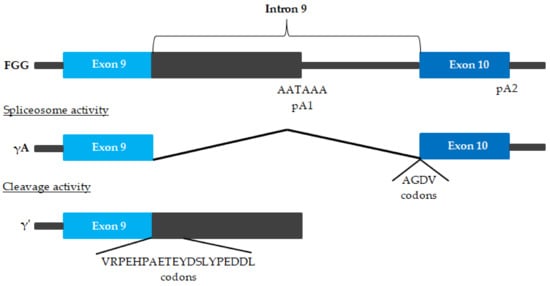
An alternatively-spliced form of the fibrinogen is γ′ fibrinogen (
Figure 3)
[14][15][16][67,68,69].
Figure 3. Alternative FGG pre-mRNA processing
[14][15][16][67,68,69]. The γA chain arises when polyadenylation occurs at polyadenylation signal 2 (pA2) downstream of exon 10, and all 9 introns are removed. The alternative chain arises after polyadenylation at pA1 in intron 9. This leads to the translation of a polypeptide with a unique 20-amino acid extension encoded by intron 9, substituting the 4 γA amino acids of exon 10.
Fibrinogen is a disulfide-bonded dimer, with each half of the dimer containing one Aα chain, one Bβ chain, and one γ chain, which can be either the more common γA chain or a γ′ chain. γ′ fibrinogen consists of approximately 90% heterodimers containing one γ′ chain and one γA chain, and about 10% homodimers containing two γ′ chains; γ′ fibrinogen constitutes about 3–40% of total fibrinogen plasma concentrations
[17][18][70,71]. In particular, compared to the total fibrinogen, γ′ fibrinogen forms fibrin blood clots that show differences in clot architecture, are mechanically stiffer, and are very resistant to fibrinolysis
[14][67].
Therefore, whether or not γ′ fibrinogen is simply a marker of CVD or a prospectively defined risk factor for CVD remains controversial. It is known that other factors also influence the structure of a blood clot. Clot structure may contribute to an increased CVD risk in vivo through associations with other CVD risk factors (age, metabolic syndrome, CRP, high density lipoprotein HDL cholesterol and homocysteine) independent from total or γ ‘fibrinogen plasma concentrationa
[19][78]. Similar conclusions were reached by Pieters et al., who stated that CVD risk factors (excluding fibrinogen) explained 20% and 3%, respectively, of the variance in fibrinogen γ′ and the γ/total fibrinogen ratio, with C-reactive protein making the biggest contribution. More than 50% of the variance in fibrinogen γ′ and γ′/total fibrinogen ratio is explained by factors other than total fibrinogen or other traditional CVD risk factors
[20][79]. Recently, Rautenbach et al. found that the iron metabolism activity may affect the plasma fibrinogen concentration and the percentage of γ′ fibrinogen
[21][80]. Moreover, another study by Rautenbach et al. also found that alcohol intake influenced the percentage of γ′ fibrinogen, as well as modulated the influence of fibrinogen SNPs on total fibrinogen plasma concentrations
[22][81]. The Atherosclerosis Risk in Communities (ARIC) study by Appiah et al. found that γ′ fibrinogen plasma concentrations seemed to reflect the general inflammation that accompanies and may contribute to ASCVD, instead of γ′ fibrinogen being a causal risk factor
[17][70]. The protective effect of γ′ fibrinogen described in some studies may be due to the different roles of fibrinogen, aside from the formation of fibrin clots, in thrombotic diseases of various aetiologies
[23][77]. The results of in vivo studies indicate that the elevated levels of the γA/γA fibrinogen isoform promote arterial thrombosis in vivo, whereas the γA/γ′ isoform does not
[24][82]. Moreover, a significant influence on the relationship between γ′ fibrinogen and the risk of CVD may depend on the presence of various polymorphisms, such as
FGG 9340T and
FGA 2224G, for example
[25][74]. In conclusion, the question of the influence of the isoform of γ′ fibrinogen on the risk of CVD requires further research.
The issue of the influence of the
FGB polymorphism on the risk of lower extremity deep venous thrombosis (LEDVT) is also interesting. In a study by Han et al. involving 120 LEDVT patients and 120 healthy people, the relationship between six SNPs in the
FGB promoter was assessed: -148 C/T, -249 C/T, -455 G/A, -854 G/A, -993 C/T and -1420 G/A and the risk of LEDVT. A higher fibrinogen plasma concentration was shown to increase the risk of LEDVT. The risk of LEDVT increased by 4.579 times for every unit increase in fibrinogen plasma concentration. It was found that polymorphisms such as -1420 (AG + AA) and -148 (CT + TT) were associated with a higher risk of LEDVT
[26][95].
It is worth mentioning that congenital fibrinogen deficiencies are rare bleeding disorders characterized by extensive genetic heterogeneity in all the three genes:
FGA,
FGB, and
FGG (encoding the Aα, Bβ and γ chain, respectively). Depending on the type and site of mutations, congenital defects of fibrinogen can result in variable clinical manifestations, which range from asymptomatic conditions to the life-threatening bleeding or even thromboembolic events
[27][28][29][96,97,98].
The influence of the more important mutations and polymorphisms of fibrinogen genes on the risk of CVD is summarized in
Table 23.
Table 23. Fibrinogen molecular modifications and cardiovascular risk. CVD - cardiovascular disease; CAD—Coronary artery disease; MACE—major adverse cardiovascular events.
| Gene |
Polymorphism/Mutation |
Effect on Cardiovascular Risk |
Bibliography |
| FGG |
γ′ fibrinogen |
↑ myocardial infraction
↑/↔ CVD
↓ venous thromboembolism and ischemic stroke |
[14][[23] | [67 | 25][30] | ,74,76,77] |
| rs7681423 and rs1049636 |
↔ CVD |
[14] | [67] |
| rs2066865 |
↑ microvascular thrombosis |
[31] | [75] |
| FGB |
-455 G/A |
↔ venous thromboembolism
↓ venous thromboembolism (Caucasians)
↑ ischemic stroke (Asian)
↔ ischemic stroke (Caucasians and children)
↑ cerebral infarction
↑ CAD
↑ cardioembolic stroke |
[32][33] | 85 | [34][35] | ,86 | [36] | ,87 | [37] | [84,,88,89] |
| -148 C/T |
↔ venous thromboembolism
↑ ischemic stroke (Asians and Caucasians)
↑ cerebral infarction
↑ CAD
↑ MACE |
[32][33][34] | [84,85,86] |
| -1420 (AG + AA) and -148 (CT + TT) |
↑ lower extremity deep venous thrombosis |
[26] | [95] |
In conclusion, the influence of molecular modifications of fibrinogen, especially the polymorphisms of its genes, on the risk of CVD remains inconsistent. One possible explanation for the discrepancy in test results may be the complexity of the regulation of fibrinogen phenotype formation. A study by Cronjé et al. found that, apart from SNPs in the fibrinogen (
FGA,
FGB and
FGG) genes, the fibrinogen phenotypes were also associated with SNPs in genes playing a role in lipid homeostasis (
LDL-R,
PCSK-9), together with
CBS and
CRP polymorphisms (particularly, CRP-rs3093068)
[38][99]. Cronjé et al. also indicated the significant effect of IL-6 on the plasma fibrinogen concentration and the properties of the clot. Moreover, they highlight the possible interactions with modulating factors and that SNP effects seem to be additive and should be taken into account
[39][100]. Similar conclusions were reached by Titov et al., who found associations between ischemic stroke and allele/genotype combinations of genes
IL6, FGA and
FGB, in which
IL6 plays key role and
FGA and
FGB have a modulating function
[40][101]. A Mendelian randomization study by Ward-Caviness et al. found a small causal effect of fibrinogen on CAD
[41][102]. The influence of molecular modifications of fibrinogen and the polymorphisms of its genes requires further research. Importantly, from a clinical point of view, polymorphisms of fibrinogen genes may modulate the effectiveness of CVD treatment.