Extracellular matrix (ECM) undergoes remodeling processes to regulate vascular smooth muscle and endothelial cells’ proliferation, differentiation, and adhesion. Abnormalities affecting the ECM can lead to alteration in cellular behavior and from this, this can conduce to the development of pathologies. Metalloproteases play a key role in maintaining the homeostasis of ECM by mediating the cleavage of different ECM components. There are different types of metalloproteases: matrix metalloproteinases (MMPs), disintegrin and metalloproteinases (ADAMs), and ADAMs with thrombospondin motifs (ADAMTSs). ADAMTSs have been found to participate in cardiovascular physiology and diseases and specifically in aortic aneurysms. This entryreview aims to decipher the potential role of ADAMTS proteins in the physiopathologic development of Thoracic Aortic Aneurysms (TAA) and Abdominal Aortic Aneurysms (AAA).
1. Introduction
An aortic aneurysm (AA) is a dilatation that occurs in the aorta, a major blood vessel that comes out of the heart and carries blood throughout the body. Two types of aortic aneurysms can transpire: (1) abdominal aortic aneurysms (AAA), which occur in the descending aorta in the abdomen, and (2) thoracic aortic aneurysms (TAA) which occur in the aortic section of the chest cavity (ascending, cross, and early descending).
Beyond cellular components such as smooth muscle cells, fibroblasts, endothelial cells, and immune cells, the aortic extracellular matrix (ECM) has a crucial role in maintaining homeostasis and the physiopathological mechanisms of thoracic aortic aneurysms and dissections (TAAD). Almost all the thickness of the aorta is made up of ECM proteins, such as fibrillar proteins (e.g., collagens, fibrillins, and elastin), proteoglycans (e.g., heparan-sulfates glycoproteins and perlecan), and metalloproteases. ECM proteins, components of vessel walls, are in permanent replacement due to a consistent turnover of proteins with the help of metalloproteases, specifically matrix metalloproteases (MMP) and a disintegrin and metalloproteases with thrombospondin motifs (ADAMTS). The hallmarks of TAA are the progressive degradation of the media due to the decreased integrity of elastic fibers, increased elastolysis activity (through metalloproteases), and TGF-β signaling pathway overactivation [1].
ADAMTS Proteins
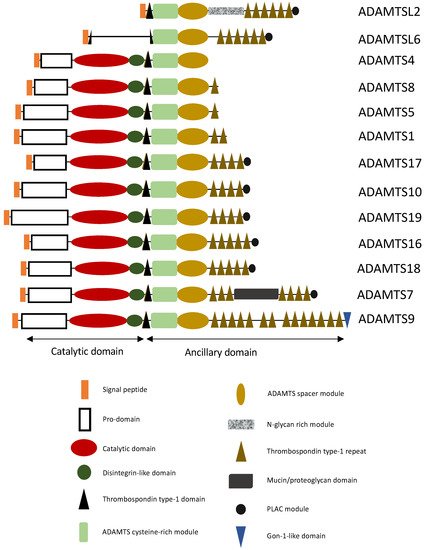
The ADAMTS family constitutes a group of proteins composed of 19 enzymes and 7 ADAMTS-like proteins. The ADAMTS share a specific domain organization with a signal peptide, a prodomain, a catalytic domain, and an ancillary domain. Their catalytic activity involves zinc and three conserved histidine residues
[2]. The ancillary domain may consist of a disintegrin-like domain, a first thrombospondin-type I motif, a cysteine-rich domain, a spacer domain, and other thrombospondin motifs. Certain ADAMTS have additional domains, such as the PLAC (protease and lacunin) domain, present in ADAMTS2, 3, 6, 7, 10, 12, 14, 16, 17, 18, and 19, a Gon-1-like domain, found only in ADAMTS9 and 20, and a mucin domain, specific to ADAMTS7 and 12 (
Figure 1).
Figure 1. Structural representation of ADAMTS(-L) involved in aortic aneurysms. ADAMTS have a characteristic domain organization consisting of a catalytic domain at their N-terminus, and a C-terminal ancillary domain. The ancillary domain has a modular organization containing distinct modules, such as an ADAMTS cysteine-rich module, an ADAMTS spacer module, thrombospondin type-1 domains and the presence or absence of PLAC module and/or mucin/proteoglycan domain (see Figure).
To be active, the zymogen form of ADAMTS proteases has an N-terminal propeptide, which must be cleaved by proprotein convertases such as furin
[3]. However, there are exceptions to ADAMTS9 and ADAMTS13, which stay active despite retention of the propeptide
[4][5][4,5]. The propeptide cleavage may occur in the trans-Golgi network, or at the cell surface
[6][7][6,7]. Most proteases of the ADAMTS family are modified by post-translational modifications, such as N- and O-glycosylation, the addition of chondroitin–sulfate chains, C-mannosylation of tryptophane residues, and O-fucosylation of serine/threonine residues in the thrombospondin type I repeat (TSR)
[2]. These enzymes’ activities are mainly regulated by inhibitors, TIMP-3, α2-macroglobulin, and endocytosis mediated by the LRP-1 receptor leading to a decrease of functional and bioavailable ADAMTS in the cellular microenvironment
[8][9][10][11][8,9,10,11].
2. ADAMTS/ADAMTSL and TAA
2.1.
THSD4
/ADAMTSL6 Mutations Responsible for TAA
The
THSD4 gene encodes the ADAMTSL6 protein. ADAMTSL6 is known to be a microfibril-associated extracellular protein without any catalytic activity. In E16.5 mouse embryos, ADAMTSL6 proteins were observed in the dermis, perichondrium, aortic wall, and all elastic tissues
[12][21]. The ADAMTSL6 protein was also present in adult kidney artery walls and the mitral valve in the adult heart. All these data suggest that ADAMTSL6 is an ECM protein associated with elastic tissues in its fibrillary components. ADAMTSL6 interacts directly with Fibrillin-1 (FBN1) at its N-terminal region and promotes early-stage FBN1 microfibril assembly
[12][21]. This role was confirmed using transgenic mice where the
Thsd4 gene was overexpressed.
2.2. Proteoglycanases Involved in TAA
Proteoglycans are one of the major groups of ADAMTS substrates. Their composition is a core protein associated with various glycosaminoglycans. In the vasculature, the proteoglycans are mainly expressed in endothelial cells and vascular smooth muscle cells of the intima and the media of the vessel wall. Due to their position in the ECM, they are involved in cell communication, signaling, and behavior. Despite the increase of proteoglycans as a hallmark of atherosclerosis plaques, recently, it was demonstrated that enhanced aggrecan and versican are also features of aneurysms
[13][27]. The level of proteoglycans is regulated by proteoglycanases such as ADAMTS. Considering the importance of proteoglycans in the vessels, we will highlight the role of these enzymes in vascular diseases.
3. ADAMTS Linked to AAA
3.1. ADAMTS8
ADAMTS8 is an aggrecanase with low efficiency due to poor proteoglycans cleavage activity
[14][49]. To date, this enzyme has been poorly investigated. Its expression profile shows expression in the heart and lungs. Its expression in the lung was correlated to its role in pulmonary arterial hypertension (PAH)
[15][50]. In a mouse model using hypoxia to induce PAH,
ADAMTS8 was found to be enhanced in the lung, as well as in the pulmonary arterial SMC (PASMC) in PAH patients. In a vascular SMC-specific mouse model,
Adamts8ΔSM22, under hypoxia-induced pulmonary hypertension it was found that PAH was reduced. Even if ADAMTS8 is part of the extracellular matrix, it was shown that ADAMTS8 has a role in intracellular signaling using PASMCs. The proliferation of PASMC was decreased when human recombinant ADAMTS8 was deleted
[15][50]. The
ADAMTS8−/− PASMC displayed increased phosphorylation of AMP-activated protein kinase/acetyl-CoA Carboxylase signaling. ADAMTS8 also may have endothelial function
[15][50]. Secreted ADAMTS8 from PASMCs might be the mechanistic connection between PA endothelial cells and PASMCs in PAH pathogenesis.
3.2. ADAMTS9
ADAMTS9 is an actor in ciliogenesis
[16][53]. This protease is also required for normal cardiovascular development. Heterozygous
Adamts9 mice were shown to have defects in the aortic wall, valvulosinus, and valve leaflets. These mice also displayed abnormal myocardial projections and a “spongy” myocardium, consistent with non-compaction of the left ventricle. These mutant mice heart anomalies were correlated with abnormal accumulation of versican and a decrease in cleaved versican compared to WT mice. These findings suggest a potentially important role for ADAMTS9 cleavage of versican in heart development
[17][54]. In addition,
ADAMTS9 expressed in vascular SMC may be a good candidate gene responsible for hereditary TAA.
ADAMTS9 expression was identified as a marker of terminal AAA. Analysis of aortic wall tissues from patients with elective or emergency repair of ruptured AAA was used to establish the molecular changes leading up to AAA rupture
[18][55].
ADAMTS9 was found to only be upregulated in tissues from patients with an emergency repair of the abdominal aorta.
3.3. ADAMTS7
It has been suggested that ADAMTS7 directly binds to the ECM protein named cartilage oligomeric matrix protein (COMP) in cartilage. COMP has been implicated in the pathogenesis of arthritis
[19][56]. COMP was discovered to be expressed not only in skeletal tissue but also in the aorta. It has also been implicated in the attachment and haptotaxis of VSMCs
[20][57]. In addition, COMP has also been associated with human atherosclerotic lesions, which suggests its potential importance during pathological ECM remodeling and VSMC migration
[21][22][58,59]. ADAMTS7 has also been found to be involved in intimal thickening after vascular injury
[23][60]. Altogether, these findings suggested that vascular remodeling is the consequence, at least in part, of ADAMTS7-dependent COMP degradation.
4. ADAMTSL2, ADAMTS10 and 17 Possibly Involved in Aorta Pathology
Some ADAMTSL family proteins are involved in rare disorders. These rare disorders, namely Acromelic dysplasias, include, among others, the Weill-Marchesani syndrome (WMS) and Geleophysic dysplasia (GD). Mutations in
ADAMTS10 (OMIM#277600) and
ADAMTS17 (OMIM#613195) have been identified in WMS cases, and mutations in
ADAMTSL2 (OMIM#231050) were associated with GD patients. These disorders share similar clinical features, such as short stature with shortened extremities, thick skin, and restricted joint mobility. These pathologies can also lead to cardiovascular defects that may be life-threatening conditions
[24][65].
GD was first described in 1971 in two patients presenting, among others, short stature, short hands and feet, and limited joint mobility
[25][66]. Homozygosity mapping in two unrelated non-consanguineous families and four unrelated consanguineous families found
ADAMTSL2 to be the causal gene of the recessive form of GD
[26][19]. The first mutations were found either in the cysteine-rich module or in the TSR6 of the ADAMTSL2 protein
[27][67].
5. ADAMTS18 Involved in Aorta Development
The role of ADAMTS18 in vascular development was researched by using an
Adamts18 knockout mouse model
[28][88]. During the development of the mice,
Adamts18 mRNA was observed in cells surrounding the ascending aorta and carotid artery.
Adamts18 knockout mice presented malformations of the embryonic aortic arch and the carotid artery system, including disordered elastic fibers and reduced carotid blood pressure. They also presented hypoplasia of the thymus and the absence of the carotid body. These abnormalities could be explained by the deficiency of Adamts18, causing fibronectin accumulation and ECM remodeling. These phenomena would, thus, activate the Notch3 signaling pathway. This activation ends up affecting the differentiation of cranial neural crest cells into VSMC
[28][88]. The repertoire of ADAMTS18 substrates needs to be further investigated to better understand the role of this enzyme in the aortic arch and carotid artery. FBN1 may be a substrate candidate. In the
Adamts18−/−, an increased level of FBN1 was observed in a carotid artery with disorganized elastic fibers and lower blood pressure
[28][88]. It has been shown that ADAMTS18 is an actor of FBN1 assembly in bronchial cells
[29][89].
6. ADAMTS19 Implicated in Valve Development
Recent genetic data revealed a potential function of
ADAMTS19 in valve development. Loss of function
ADAMTS19 mutations have been identified as being responsible for non-syndromic heart valve disease
[30][90]. Patients with heart valve disease represent only 2% of the general population. Using whole-exome sequencing, homozygous truncating nonsense variants in
ADAMTS19 were revealed in four affected cases of two distinct families. To understand the role of ADAMTS19 in valve development, a mouse model using the knockout first allele tagged with a LacZ reporter cassette was generated. The follow-up of the echocardiographic analysis displayed a progressive aortic valve disease at three months of age in 38% of the
Adamts19KO/KO mice. The abnormal valves presented disorganization of the ECM, which could be a consequence of the mutant metalloproteinase ADAMTS19. Mutants displayed higher proteoglycan content as well as thinner collagen fibers. Using LacZ staining, the expression of
Adamts19 was strongly detected in all four valves, and specifically in the valvular interstitial cells (VICs). Single-cell transcriptomic data suggested that there is a defect in the mechano-transduction pathway from VIC to endothelial cells, implying the Wnt signaling pathway.