Outside of the Earth’s protective magnetosphere, crews are exposed to galactic cosmic rays (GCR) and solar proton events (SPE) that occur when particles emitted by the sun, mostly protons, become accelerated in the interplanetary space due to a coronal mass ejection shock
[1]. The exposure to GCR in the form of high-energy (HZE) ions, secondary protons, and neutrons has high linear energy transfer values that evoke complex DNA and other cellular damage
[2]. In the purpose of controlling the health risks associated with the unique hazards of space flight, highly sophisticated systems have been developed
[3]. A key question that impacts risk assessment is how cancers caused by HZE radiation compare to either radiogenic cancer induced by ground-based radiation
[4]. As a unifying concept, NASA studies have sought to examine how space radiation exposure induces genetic and epigenetic modifications, noted as the hallmarks of cancer onset
[5]. Due to the lack of human epidemiological data related to the types of radiation found in space, the current research utilises a translational approach which includes advanced human cell-based model systems exposed to space radiation simulants connected with human molecular pathways
[6]. This approach allows relating the biological effects of space radiation to effects from similar exposure to ground-based gamma rays and X-rays to extrapolate the results to large human epidemiological cohorts
[6][7][6,7]. Among the molecules involved at the cellular level, the CREB/ATF family plays a crucial role.
The CREB protein was initially described as a cAMP-responsive transcription factor involved in the regulation of the somatostatin gene
[8]. Today, it is known to modulate gene transcription through binding to DNA sequences known as the cAMP response elements (CREs)
[9]. In the human genome, there are approximately 750,000 of these CREs, although most of them are not available for protein binding as their cytosine methylation physically prevents these interactions
[10]. The CREB/ATF transcription factors have key roles in cell survival, proliferation, and differentiation, as well as in apoptosis and adaptive responses
[11]. Nuclear factor-κB (NF-κB) appears to be the most important CREB-related transcription factor
[12]. Increased NF-κB activity can be considered as a hallmark of different diseases, such as human leukaemia, lymphoma, and other types of cancers
[13]. NF-κB activity can be induced by ionising radiation, which appears to promote enhanced survival of human leukaemia K562 cells
[14]. Indeed, a link has been shown between constitutive NF-κB activity, basal apoptosis, and radiosensitivity in breast carcinoma cell lines
[15]. Furthermore, high NF-κB activity in human cancers can promote apoptosis suppression and radiotherapy resistance
[16].
2.1. Cell Responses to Ionising Radiation
Ionising radiation is responsible for the loss of cell proliferation and for cell death by apoptosis or necrosis. Of note, there are two types of apoptosis: fast apoptosis, which occurs during the interphase, before cell division, and after the G2 block that is induced by radiation; and late apoptosis, which occurs after one or more cell divisions
[17][110]. Some cells, such as lymphocytes, thymocytes, and intestinal crypt cells, are particularly radiosensitive, and when they are irradiated, they undergo fast apoptosis; when mouse leukaemia cells are irradiated, they experience G2 block, and mainly undergo apoptotic death
[18][111].
As a response to genomic stress, activation of p53 can result in cell cycle arrest or cell death by apoptosis, and this can also contribute to DNA repair processes
[19][112]. The mouse double minute (Mdm) 2 protein is a crucial regulator of p53. In mice, inactivation of the
mdm2 gene shows early embryonal lethality
[20][113]. Mdm2 has a dual relationship with p53. When Mdm2 binds to p53, this can inhibit the transcriptional function of p53, which also results in complete elimination of p53 by proteolytic degradation. At the same time, p53 can bind the
mdm2 gene, which stimulates its transcription. Therefore, this defines a negative feedback loop that appears to serve to rapidly terminate the p53 response after effectively dealing with the p53-activating stress signal
[21][114]. Various mechanisms have been proposed to explain the p53 fluctuations that are observed in cell populations
[22][115]. However, considering the continuous effects on cells of acute ionising radiation, the complex cell responses that can be activated to fight against DNA damage still need to be addressed further at the level of the single cell. For oncogenes and toxins that have p53 regulatory functions, their degradation kinetics can be used to quantitatively predict outcomes of the cell responses to DNA damage induced by different doses of ionising radiation
[23][116]. The retinoblastoma protein (Rb) and CBP/p300 have more complex influences on the p53/MDM2 interactions. Here, the binding of Rb to MDM2 prevents the MDM2 destabilisation of p53 while the Rb/MDM2 complex continues to bind to p53 and inhibits the transactivation mediated by p53
[24][117]. Thus, this indicates how the p53 inhibition and destabilisation functions of MDM2 can be individually deciphered. Regarding CBP/p300, these coactivators of p53 are also required for p53 degradation mediated by MDM2. When MDM2 lacks its p300-binding domain, it can no longer destabilise p53, although the MDM2 and p53 binding appears not to be affected
[25][118].
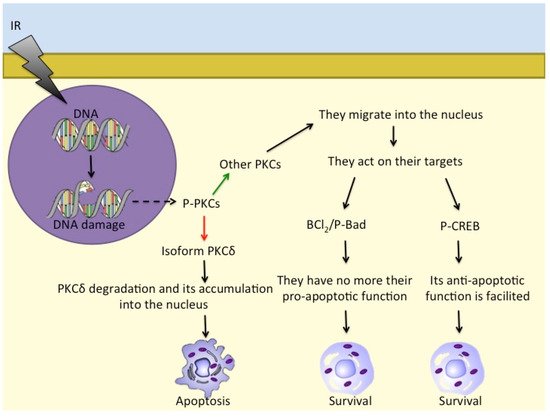
There are further proteins that are involved in cell responses to ionising radiation, such as PKC family members
[26][27][92,119]. PKCs are serine/threonine kinases and comprise (at least) 12 different isozymes. PKCδ releases a 40 kDa fragment by proteolysis when cells are exposed to ionising radiation and also to DNA-damaging drugs that leads to apoptosis
[28][120]. Furthermore, in response to irradiation, activated PKCs regulate P-Bad, Bcl-2, and CREB to prevent apoptosis and induce pro-survival signalling
[29][121]. PKCs also translocate into the nucleus to phosphorylate its targets
[30][31][122,123] (
Figure 12).
Figure 1. Different PKC isoforms respond to DNA damage in different ways: while PKCδ accumulates in the nucleus and induces apoptosis, other PKCs act on Bcl2/P-Bad and on P-CREB to promote cell survival. - - -→ = response to DNA damage; ––> = activation; ––> = inhibition; ––> = consequentiality.
2.2. CREB and Other Factors in Radioresistance and Radiosensitivity
Radiotherapy is designed to induce DNA double-strand breaks, which would then lead to elimination of cancer cells via apoptosis
[32][124]. However, the efficacy of radiotherapy treatment against cancers also depends on the toxic side effects, which can impede dose escalation. Furthermore, as indicated above, cancer cells might instead develop radioresistance through mechanisms related to DNA repair responses. When cells are exposed to ionising radiation, there is activation of transcription factors like AP-1 and NF-κB
[33][125]. The consequent induction of specific genes and synthesis of their protein products might then provide the cells with radiation resistance. When the constitutive levels of NF-κB are high, cells also show relatively high resistance to radiation therapy
[34][126]. The Daudi and Ramos cell lines (human Burkitt lymphoma) show sensitivity to relatively low radiation doses (i.e., 1–5 Gy), with reduced cell viability due to necrosis and apoptosis, and the cell cycle blocked in the G
2/M phase
[35][93]. The less radiosensitive Ramos cells show expression of a mutated form of p53 and a constitutively activated NF-κB pathway. These cells are sensitive to an ionising radiation dose of 3 Gy, where they show an early increase in the expression of CREB and a dose-dependent upregulation of expression of NF-κB
[35][93]. Interestingly, increased cellular levels of CREB have proapoptotic effects, while NF-κB upregulation can be linked to necrosis, at least in vitro and after 3 Gy ionising radiation
[35][93].
Concerning the regulation of NF-κB, normally, its nuclear translocation and activation are prevented by the “super-repressor” IκB, but stimuli such as TNFα and ionising radiation can degrade, and thus remove, IκB, leaving NF-κB free to translocate into the nucleus and activate its target genes
[36][127]. Therefore, as NF-κB is activated in several types of cancer, this might provide the cells with intrinsic radioresistance or promote radioresistance. Indeed, NF-κB activity induced by ionising radiation appears to enhance the survival of K562 human leukaemia cells
[14]. Furthermore, in breast carcinoma cell lines, a link has been reported for constitutive NF-κB activity, basal apoptosis, and radiosensitivity
[37][128]. This indicates that the higher levels of NF-κB seen for human tumours can both suppress apoptosis and promote radioresistance. Inhibition of the NF-κB expression generally increases the apoptotic response when cells are under radiation therapy
[38][129], and as indicated above, NF-κB expression is upregulated in certain tumour cells in response to radiation, and to chemotherapeutic drugs
[39][130]. Instead, K562 leukaemia cells have shown a different strategy for resistance to apoptosis induced by ionising radiation that is modulated through protein kinase C (PKC) δ and NF-κB
[14].
CREB has an active role in prostate cancer, where radiation therapy is the first-line treatment
[40][131]. In the human prostate, neuroendocrine cells represent one of three types of epithelial cells
[41][132]. These neuroendocrine cells can promote growth of the surrounding tumour cells through their release of neuropeptides
[42][133]. Along with CREB, a role for ATF2 has been indicated in prostate cancer. Indeed, it has been hypothesised that ATF2 acts as a shuttling protein as it moves between the cytoplasm and the nucleus
[43][134]. Ionising radiation can induce reversible differentiation of neuroendocrine cells, which leads to the loss of their neuroendocrine properties. Here, CREB and ATF2 might have opposing effects as it appears that accumulation of ATF2 in the nucleus antagonises the signalling pathway involved in the phosphorylation of CREB that is induced by ionising radiation. After exposure to ionising radiation, the differentiated cells show increased proliferation, thus losing their neuroendocrine-like properties
[44][135]. In this case, radiotherapy also gives tumour cells the possibility to survive the treatment and contribute to tumour recurrence.
Radioprotection of human leukaemia cell lines can also be linked to the stability of peroxiredoxins (Prx)
[45][136]. They are a very large and conserved family of small peroxidases that conserve the thioredoxin-dependent catalytic activity that protects cells from oxidative damage produced by H
2O
2, organic hydroperoxides, and peroxynitrite
[46][137]. The effects of ionising radiation in leukemia cells are of oncologic interest since high doses of whole-body gamma radiation can be employed before bone marrow transplantation. However, PrxII expression levels have been correlated with radioresistance or administration of certain anticancer drugs in radioresistant solid tumours, including breast cancer, glioblastoma, and head and neck cancer, as well as in tissue isolated from head and neck patients who do not respond to radiation therapy
[47][138].
2.3. CREB and Related Transcription Factors as Possible Targets in the Treatment of Tumours
CREB appears to be a therapeutic target for cancer treatment due to its role in the development, maintenance, and progression of tumours
[48][94]. This was also suggested by the downregulation of the inducible cyclic AMP early repressor (ICER) in bone marrow cells from patients with acute myeloid leukaemia, where altered CREB expression was observed. There are several different ways in which the CREB function might be inhibited in tumour cells
[48][94]. First, a dominant-negative CREB mutant, known as KCREB, can be used to inhibit transcription of CREB, which arises through the formation of heterodimers of KCREB with wild-type CREB. In metastatic tumour cells in vitro and in vivo, KCREB has been shown to reduce the metastatic potential
[49][95]. Secondly, CRE “decoy” oligonucleotides might inhibit
CREB gene transcription and tumour growth
[50][139]. Thirdly, silencing of CREB expression might reduce anchorage-independent growth of tumour cells, along with cell cycle arrest. This would lead to apoptosis accompanied by enhanced tumour immunogenicity
[51][140]. Finally, many kinase inhibitors can be used to inhibit the CREB and CBP interactions with their CREs
[52][141].
Pharmacological inhibition of IKK-NF-κB might also represent an interesting approach to potentiate the apoptotic effects of irradiation. Indeed, a suppressor of the IKK complex was reported to make lung cancer cell lines more apoptosis-susceptible following their irradiation
[53][142]. Furthermore, curcumin, which interferes with activation of the inhibitor of NF-κB kinase (IKK), showed greater radiation-induced apoptosis against PC-3 prostate cancer cells
[54][143]. Instead of targeting NF-κB itself, the effector genes of NF-κB might represent better drug targets for the enhancement of radiosensitivity on the basis that in tumour cells that are radioresistant, activation of these genes might be predominant. In an investigation of genes that are upregulated in human keratinocytes following long-term irradiation, Chen et al. (2002)
[19][112] reported that six genes showed upregulation as putative targets for NF-κB. By using a mutant IκB, three of these genes, cyclin B1, cyclin D1, and human inhibitor of apoptosis protein (HIAP) 1, were downregulated, while the other three upregulated genes, Bcl-2-associated athanogene (BAG) 1, thyroid transcription factor (TTF), and fibronectin, were not downregulated. The genes that were downregulated following NF-κB inhibition are associated with significantly decreased cell survival, and thus they might have crucial roles in radioresistance.
For hepatocyte malignancy in HCC, radiotherapy is the most common treatment choice
[55][144]. Unfortunately, as both cancer and healthy cells are killed, this treatment comes with multiple side effects for the cells. However, as Fuchs-Tarlovsky (2013)
[56][145] reported, administration of antioxidant nutrients prior to or combined with radiation therapy protected nontumour cells against the free radicals generated to kill the tumour cells during the irradiation.
P53 can also be considered as a target in anticancer therapies that use different small molecules that have been shown to restore the function of wild type p53. One of these is the cis-imidazoline analogue nutlin-3 which prevents p53 degradation and was shown to induce apoptosis in p53-deficient colorectal carcinoma cells and in an HCC cell line through p73 activation
[57][58][146,147]. Previous preclinical studies showed the therapeutic use of nutlin-3 for haematological malignancies, including acute myeloid leukaemia
[59][148], acute lymphoblastic leukaemia
[60][149], and B cell chronic lymphocytic leukaemia
[61][150]. In a human head and neck cancer cell line, the small molecule known as RITA (reactivation of p53 and induction of tumour cell apoptosis) that blocks the interaction between p53 and MDM2 was shown to restore the function of p53 and induce tumour cell apoptosis
[62][151]. Indeed, there are several other small molecules that are used in therapies for tumours where p53 mutations have led to loss of the p53 DNA-binding function. For example, PRIMA (p53 reactivation and induction of massive apoptosis) 1 reactivates p53 and induces apoptosis
[63][152]. Then, a small molecule with a similar structure to PRIMA-1 known as PRIMA
MET (APR-246) can also induce tumour cell apoptosis either alone or in combination with other chemotherapeutics. Furthermore, in multiple myeloma, MIRA (mutant p53 reactivation and induction of rapid apoptosis) 1 can restore the p53 function and p53-induced cancer cell apoptosis with a higher potency than PRIMA-1
[64][153]. Then, there is the small molecule RETRA (reactivation of transcriptional reporter activity) which not only restores the p53 function, but also increases the level of Tap73, a structural homologue of p53. However, the essential problem with these small molecules is the need for selective actions (in terms of restoration of the p53 function) that are directed only to the cancer cell.