Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Masfique Mehedi and Version 2 by Peter Tang.
Respiratory syncytial virus (RSV) is the leading viral agent causing bronchiolitis and pneumonia in children under five years old worldwide. The RSV infection cycle starts with macropinocytosis-based entry into the host airway epithelial cell membrane, followed by virus transcription, replication, assembly, budding, and spread. It is not surprising that the host actin cytoskeleton contributes to different stages of the RSV replication cycle. RSV modulates actin-related protein 2/3 (ARP2/3) complex-driven actin polymerization for a robust filopodia induction on the infected lung epithelial A549 cells, which contributes to the virus’s budding, and cell-to-cell spread.
- cytoskeleton dynamics
- filopodia
- ARP2/3 complex
- actin polymerization
- cell-to-cell spread
- RSV
- bronchiolitis
- therapeutics
1. Introduction
Microorganisms, including viruses, use the host cell’s cytoskeleton to destabilize the host cell’s physiological mechanisms to allow for the virus’s survival and aid its pathogenesis. Most bacteria and viruses utilize the host cytoskeleton for multiple activities, including attachment, invasion, movement within and between cells, and replication, resulting in disease progression [1][2][3][1,2,3]. Actin microfilaments are unique among the cellular cytoskeletons, as they are composed of a highly dynamic network of actin polymers. Host cells contain actin-associated proteins that modulate cell migration, contraction, and shape changes during the cell cycle and in response to extracellular stimuli [4][5][6][4,5,6]. During a microbial attack, the induction of macropinocytosis, phagocytosis, membrane ruffling, vacuole formation, and vacuole remodeling depends on signaling the actin cytoskeleton [4][5][6][4,5,6]. Pathogens, like viruses and bacteria, have different strategies to hijack the host cell machinery to promote their replicative cycles; specifically, the host cytoskeleton is their common target [3][4][3,4].
The ARP2/3 complex, an actin filament nucleating and regulating factor, plays a central role in cellular actin assembly. The complex is an assembly of seven proteins, including actin-related proteins ARP2, ARP3, and five additional subunits called actin-related protein 2/3 complex (ARPC), including p41, p34, p21, p20, and p16 (noted as ARPC 1–5, respectively) (Figure 1) [7].
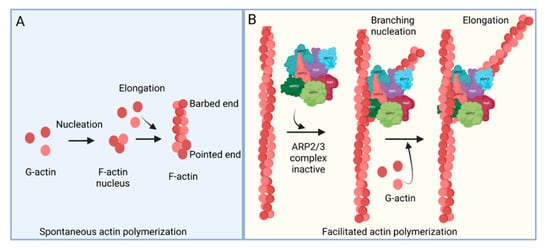
Figure 1. Actin polymerization. (A) Spontaneous actin polymerization. Globular actins (G-actin) form follicular actin (F-actin) nucleus (shown as pointed end). Spontaneous addition of G-actin elongates F-actin at the barbed end. (B) Facilitated actin polymerization. ARP2/3 complex, a seven-protein complex, consists of ARP2, ARP3, ARPC1, ARPC2, ARPC3, ARPC4, and ARPC5. ARP2/3 complex involves branched actin polymerization [8][22].
Respiratory syncytial virus (RSV) causes severe lower respiratory illnesses, such as bronchiolitis and pneumonia, in children under the age of five. Elderly adults, as well as adults with chronic diseases, are at an increased risk for contracting a severe illness from an RSV infection. Importantly, almost everyone has been infected by RSV by the time they reach two years of age. RSV infection primarily causes common cold-like symptoms that progress to lower respiratory tract disease in 40 percent of infected infants [9][23]. RSV belongs to the Pneumoviridae virus family. It is a negative-sense, single-stranded, non-segmented RNA virus. Its genome consists of 10 genes that encode 11 proteins. The proteins encoded by RSV are nonstructural protein 1(NS1), NS2, nucleoprotein (N), phosphoprotein (P), matrix (M), short hydrophobic (SH), glycoprotein (G), fusion (F), M2-1 and M2-2, and RNA-dependent RNA polymerase (L) [10][24]. The two main proteins that are widely studied for anti-viral drug discovery are F and G protein. F protein is a surface glycoprotein that is involved in the RSV infection. G protein attaches to the host cell receptor [11][25]. F protein enables the virion membrane to fuse with the host cell membrane [12][26]. Upon entering the host cell, RSV undergoes transcription, translation, and replication in the host cytoplasm [10][24]. During transcription, the viral polymerase starts mRNA synthesis for all genes from 3′ to 5′ end of the genome [13][27]. Importantly, RSV 3′ to 5′ end genes undergo a higher to lower transcription gradient [13][14][27,28]. All protein-specific mRNAs are translated by host cell translational machinery [10][24]. N protein is involved in creating a template for RNA synthesis and P protein is a polymerase co-factor [10][24]. M protein is involved in the inhibition of host transcription and is associated with viral inclusion bodies [15][29]. M2-1 is a transcription processivity factor and M2-2 regulates RNA synthesis [16][17][30,31]. NS1 and NS2 proteins are non-structural proteins that are involved in various processes including interfering with the innate immune response and inhibiting apoptosis [18][19][32,33]. SH protein is a transmembrane protein near the N-terminus involved in various processes including the inhibition of TNF signaling and reducing apoptosis [20][34]. The virus is assembled at the cell surface with viral proteins and genomic RNA. The assembled new virion budded out from the surface of the cell [10][24]. RSV attachment to cell membrane activated various signaling cascades like Epidermal growth factor (EGF), cell division protein 42 (Cdc42) which led to actin rearrangement and increased fluid uptake which results in RSV uptake by macropinocytosis, as shown in Figure 2. Actin rearrangement plays an important role in RSV entry, as treatment with cytochalasin D and latrunculin A disrupt actin filament and reduce RSV infection in HeLa cells [21][35]. Previously, it has also shown that the cytoskeleton protein actin is involved in RSV endocytosis, replication, gene expression, and morphogenesis (Figure 2) [22][23][24][25][36,37,38,39]. It has recently been shown that ARP2/3 and virus-induced filopodia contribute to RSV cell-to-cell spread (Figure 2 and Figure 3) [26][27][28][40,41,42].
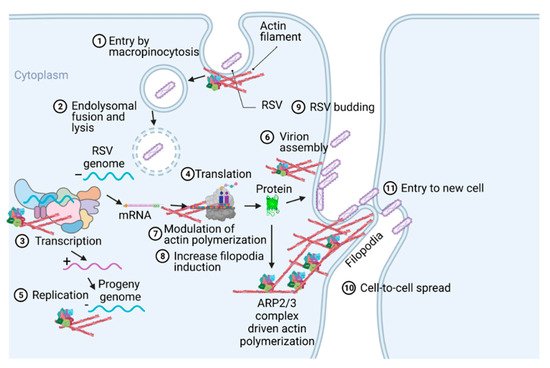
Figure 2. A pictogram of a replicative cycle of RSV. Different steps of RSV replicative cycle (including potential actin involvement) are indicated chronologically.
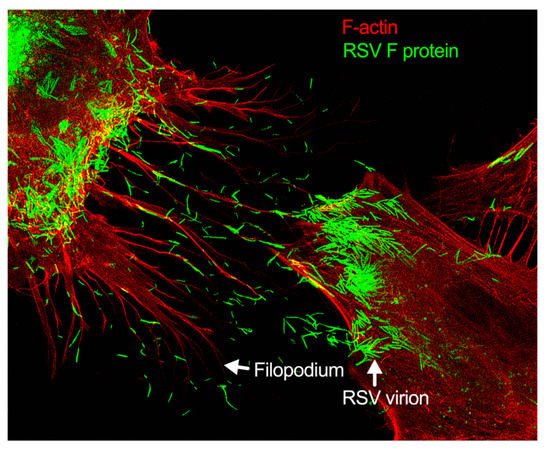
Figure 3. Filopodia-driven RSV cell-to-cell spread. Human lung airway epithelial cell line, A549 cells, were infected with RSV wild type (RSV-WT) (Strain A) at a multiplicity of infection (MOI) of 1 for 24 h. The infected cells were then fixed, permeabilized, and stained for RSV fusion (F) protein using antibody specific to F. F-actin and the nucleus were stained with rhodamine phalloidin and DAPI, respectively. The image was taken under a stimulated emission depletion (STED) microscope (Leica Microsystem) [27][28][41,42].
2. Pathogen-Induced Subversion of Cytoskeleton
The actin cytoskeleton is critically important to maintaining cellular morphology and motility. Pathogens co-opt the actin restructuring machinery of the host cell to access or create a favorable environment for their replication. Viruses are known to stimulate a rearrangement of the host cell actin cytoskeleton during the infection of the cell [29][43]. The actin cytoskeleton can control endocytosis and phagocytosis, as well as cell contraction, motility, and division [30][44]. The cytoskeleton’s capacity to continuously assemble and disassemble allows it to take on many roles [29][43]. Pathogens manipulate the cytoskeleton to drive cellular infection due to its role of being a major host structural component [31][45]. A pathogen will utilize effector proteins to hijack the cytoskeleton to successfully infect and replicate [31][45]. For example, actin reorganization is stimulated during microbial entry by binding with CR3 receptors in macrophage [32][46]. In contrast, translocated actin recruiting phosphoprotein (TARP) in non-phagocytic cell facilitates entry by promoting actin nucleation [33][47]. Cortactin, which is an actin regulatory protein, is also important for the entry in non-phagocytic cells [34][48]. ARP2/3 complex regulation by Src and PI-3 kinase were shown in bacterial internalization [35][49]. Actin-mediated propulsion is also necessary for cell-to-cell spread but not always dependent on N-WASP and ARP2/3 complex [36][50]. The ARP2/3 complex, as well as pathogen-induced filopodia, are used as mechanisms to subvert the cytoskeleton by stimulating actin assembly in the host cell [37][51]. The mechanism of the ARP2/3 complex is utilized in the formation of branched actin, while the activation of Cdc42 plays a role in the development and formation of filopodium [38][52]. Thus, various pathogens exploit the host cell actin cytoskeleton by implementing mechanisms to subvert it [37][51].
3. Role of Cytoskeleton in RSV Infection Cycle
The host cytoskeleton is involved in various stages of the RSV infectious cycle (Figure 2 and Figure 3). RSV interacts with multiple cellular and cytoskeletal proteins during infection. Nucleolin located in the apical cell surface interacts with RSV glycoproteins which is important for RSV entry [39][61]. The cytoskeletal proteins, e.g., actin and microtubules, are implicated in the RSV transcription, assembly, and budding [40][62]. F protein cytoplasmic tail and M protein are responsible for viral assembly, specifically, F protein interacts with inclusion bodies which in turn facilitate the release of M protein-ribonucleoprotein complex from inclusion bodies [41][63]. RSV M protein interacts with actin which is important for budding and responsible for the virion particle transport [42][64]. Cytochalasin D inhibits actin polymerization, which reduces the production of viral particles [23][37]. RSV also increases the polymerization of actin, which results in cytoplasmic extension in the infected cells [22][36]. Profilin, an actin modulatory protein, is also essential for RSV transcription [43][65]. Apart from actin and profilin, actin-associated signaling pathways like RhoA, PI3K, and Rac GTPase are involved in the production of virus filaments [44][45][46][66,67,68]. Microtubules play a role in both assembly and release of the virus but have a greater impact on the assembly [25][39]. Higher expression of RhoA and pMLC2 were observed during RSV infection which induced stress fiber formation and ROCK inhibitor Y-27632 showed the inhibition of RhoA activity [47][69].
Generally, respiratory viruses enter the human via the lung epithelia. This can prove challenging, however, as most of the lung epithelia are lined with a protective coating of mucus. The few cells that are not lined with mucus are guarded by macrophages. This has made particle release, the common method of viral spread, disadvantageous for these types of viruses. To avoid these pitfalls, respiratory viruses have most likely evolved novel mechanisms of spreading cell-to-cell. RSV modulates cytoskeletal rearrangement and actin remodeling for its replicative cycle in vitro [25][39], particularly ARP2/3 complex-regulated filopodia formation for its cell-to-cell spread [26][27][40,41]. When A549 cells are infected with RSV, the cell forms an actin-based projection called filopodia (Figure 2 and Figure 3) [26][27][40,41]. Filopodia are finger-like projections comprised of polymerized actin. The free barbed ends of the actin filament can add additional actin monomers, which leads to actin polymerization [48][75]. The formation of the filopodia is activated by Cdc42, a protein involved in the regulation of proliferation in the cell division cycle [49][50][51][52][76,77,78,79].
4. Role of Filopodia in RSV Cell-to-Cell Spread
Filopodia are typically involved in activities such as migration and wound healing. Sites of filopodia formation tend to have increased amounts of the protein katanin [53][80]. Katanin, in these increased amounts, has been linked to more aggressive migratory behaviors in prostate cancer cells [54][81]. Cells using directional migration also use their filopodia to establish a single polarity axis. The cell’s filopodium serve as the leading process for directional migration [55][56][57][82,83,84]. Filopodia can form multiple protrusions branching out from the filopodia (or the lamellipodia), and these branches are often able to be used by the cell to make directional decisions in chemotaxis and pathfinding [58][59][85,86]. However, herein lies the difference between the cell’s filopodium and the cell’s lamellipodium: the role of the lamellipodium tends to be more reserved for migratory purposes. The thin sheet protrusion pulls the cell forward. In contrast, filopodia play a role that is less focused on pulling the cell forward but more so on exploring the extracellular environment. The filopodia are less sheet-like and more finger-like [60][61][87,88]. In certain cells, filopodia may also play a role in phagocytosis. For example, in certain non-polarized cell types, such as primary human corneal fibroblasts, or a cancerous cell line Chinese hamster ovary (CHO) cells stably expressing nectin-1, HSV-1 entry occurs through a mechanism that seems to be “phagocytosis-like”. It includes filopodia-like actin rearrangements and RhoA GTPase activation [62][89]. Conversely, Clement et al. also showed that two actin-depolymerizing agents—CytoD and latrunculin B (LatB)—are potent inhibitors of this HSV-1 entry in these cell types. Further studies in polarized retinal pigment epithelial (RPE) cells, which have a natural tropism for HSV-1 infection, also revealed the role of actin in facilitating virus entry [62][89].