SALL proteins are a family of four conserved C2H2 zinc finger transcription factors that play critical roles in organogenesis during embryonic development. They regulate cell proliferation, survival, migration, and stemness; consequently, they are involved in various human genetic disorders and cancer. SALL4 is a well-recognized oncogene; however, SALL1–3 play dual roles depending on the cancer context and stage of the disease.
- SALL1
- SALL2
- SALL3
- SALL4
- cancer
- epigenetic regulation
- Wnt
- PTEN
- biomarker
1. Introduction
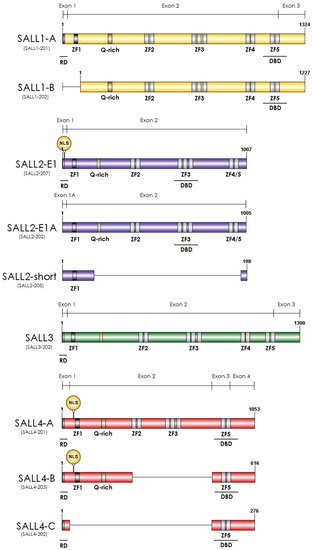
2. Essential Roles of SALL Genes during Development
3. Common Cellular Functions and Targets of the SALL Proteins in Cancer
3.1. Cell Proliferation
3.2. Apoptosis and Cell Survival
3.3. Cell Migration and Invasion
3.4. Stemness
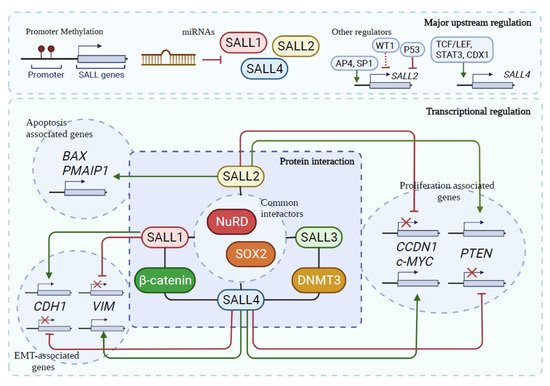
4. Common Regulatory Mechanisms for SALL Proteins in Cancer
Cancer Type/Cellular Model | microRNA | Target | SALL Status/Key Findings | Experimental Approach | Ref. | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Glioma/Glioblastoma | miR-302/367 cluster | SALL2 | miR-302/367 cluster can reprogram tumor cells, generating more benign phenotypes by suppressing OCT3/4, SOX2, KLF4, c-MYC, POU3F2, OLIG2, and SALL2 | Undescribed | qRT-PCR, cytokine array analysis | Expression correlated with lower overall survival of NSCLC patients | [94] |
[101] |
|||||||||||||||||||||||||||||
Oncogene | [ | ] |
[122] |
Glioma/Glioblastoma | miR-16 | SALL4 | miR-16 inhibits proliferation, migration, and invasion in glioma cells by directly targeting SALL4 | qRT-PCR and Luciferase reporter assay | |||||||||||||||||||||||||||||
Lung | [ | SALL2 | ] |
[102] |
|||||||||||||||||||||||||||||||||
Low | LOH | Glioma/Glioblastoma | miR-103/miR-195/miR-15-B | SALL4 | miR-103, miR-195, and miR-15-B inhibit proliferation, migration, and invasion and promote apoptosis in glioma by directly targeting SALL4 | qRT-PCR, Western blot, and Luciferase reporter assay |
[96] |
[103] |
|||||||||||||||||||||||||||||
Undescribed | Undescribed |
[64] |
[71] |
||||||||||||||||||||||||||||||||||
Lung | SALL4 | High | Undescribed | Expressed in 88% of the lung cancer samples | May be used as a diagnostic marker | Oncogene |
[116] |
[123] |
Glioma/Glioblastoma | miR-107 | SALL4 | miR-107 inhibits proliferation and promotes apoptosis in glioma cells by directly targeting SALL4 | qRT-PCR, Western blot, and Luciferase reporter assay | ||||||||||||||||||||||||
Lung | SALL4 | [ |
High | Undescribed | ] |
SALL4 knockdown inhibits cell proliferation by cell cycle arrest at the GO/G1 phase | Loss of SALL4 function inhibits migration, invasion and reduces the size of the transplanted tumor in an in vivo model. |
[104] |
|||||||||||||||||||||||||||||
Oncogene | [ | ] |
[43] |
Glioma/Glioblastoma | miR-181b | ||||||||||||||||||||||||||||||||
Lung | SALL4 | SALL4 | High | miR-181b inhibits proliferation, migration, and invasion and promotes apoptosis in glioma by directly targeting SALL4 | Undescribed | qRT-PCR, Western blot, and Luciferase reporter assay | SALL4 silencing sensitizes cells to cisplatin, carboplatin, and paclitaxel treatment |
[98] |
[105] |
||||||||||||||||||||||||||||
Oncogene | [ | ] |
[124] |
Gastric cancer | miR188-5p | SALL4 | miR-188-5p promotes proliferation and migration by upregulating SALL4 expression, nuclear translocation, and transcription | qRT-PCR, Western blot, and Luciferase reporter assay | |||||||||||||||||||||||||||||
Esophageal | [ | 99] |
[106] |
||||||||||||||||||||||||||||||||||
SALL1 | Low | Hypermethylation | SALL1, ADHFE1, EOMES, and TFPI2 are proposed as part of a tumor suppressors panel with diagnostic relevance | Tumor suppressor | Gastric cancer | miR-16 | |||||||||||||||||||||||||||||||
Esophageal | SALL2 | SALL4 | Low in radioresistant ESCC cell lines | miR-16 inhibits proliferation and migration in GC by directly targeting SALL4 | Hypermethylation | qRT-PCR and Luciferase reporter assay | SALL2 overexpression enhances apoptosis after radiation and decreases migration, viability, and cisplatin resistance in TE-1/R and Eca-109/R cell lines |
[100] |
[107] |
||||||||||||||||||||||||||||
Tumor suppressor | [ | ] |
[55] |
Colorectal cancer | miR-181a-2 * | ||||||||||||||||||||||||||||||||
Esophageal | SALL4 | SALL1 | High | miR-181a-2 * correlates with a trend of repression of SALL1 and high methylation status of the SALL1 promoter | qRT-PCR and bisulfite modification followed by quantitative methylation- specific PCR (qMSP) | Undescribed | SALL4 silencing in ESCC cells is associated with suppressing cell migration, invasion, viability, and drug resistance in vivo | SALL4 knockdown reduces epithelial-mesenchymal transition by targeting the Wnt/β-catenin signaling pathway |
[101] |
[108] |
|||||||||||||||||||||||||||
Oncogene | [ | ][119] | Colorectal cancer | miR-219-5p | |||||||||||||||||||||||||||||||||
Bladder | SALL4 | SALL2 | miR-219-5p inhibits proliferation, migration, and invasion, reduces drug resistance, and promotes apoptosis in CRC by directly targeting SALL4 | Low | LOH | qRT-PCR, Western blot, and Luciferase reporter assay | Undescribed | [102] |
[109] |
||||||||||||||||||||||||||||
Tumor suppressor | [ | ] |
[70] |
Colorectal cancer | miR-3622a-3p | ||||||||||||||||||||||||||||||||
Bladder | SALL4 | SALL3 | miR-3622a-3p inhibits proliferation, cell cycle, migration, invasion, and stemness features and promotes apoptosis by targeting SALL4 | Low | Hypermethylation | qRT-PCR, Luciferase assay, RNA immunoprecipitation (RIP) assay, and pull-down assay | SALL3, CFTR, and TWIST1 are proposed as disease recurrence predictors | [103] |
[110] |
||||||||||||||||||||||||||||
Tumor suppressor | [ | ][121] | Embryonic stem cell | miR15-B | SALL4 | Anti-miR-15b enhances expansion of HSC in vitro by targeting SALL4 | qRT-PCR | ||||||||||||||||||||||||||||||
Testicular tumors | SALL4 | High | Undescribed | SALL4 is a novel sensitive and specific marker for testicular germ cell tumors | [104] |
[111] |
|||||||||||||||||||||||||||||||
Oncogene | [ | ] |
[129] |
Embryonic stem cell | |||||||||||||||||||||||||||||||||
Kidney | miR-294 and let-7 miRNAs | SALL4 | Let-7 miR family inhibits self-renewal genes, and miR-294 indirectly induces self-renewal genes, including SALL4 | SALL1 | qRT-PCR, Western blot, and Luciferase reporter assay | Low | miR-942 | SALL1 inhibition plays a potential role in sunitinib resistance in RCC patients | [105] |
[112] |
|||||||||||||||||||||||||||
Tumor suppressor | [ | ] |
[115] |
Oral squamous cell carcinoma | |||||||||||||||||||||||||||||||||
Wilms’ tumor | miR-103 | SALL1 | SALL4 | High | miR-103 inhibits cell proliferation and invasion by downregulating SALL4 mRNA in Tca8113 cells | Undescribed | Luciferase reporter assay | Undescribed |
[106] |
[113] |
|||||||||||||||||||||||||||
Oncogene | [ | ][124] | Breast cancer | SNHG12 and miR-15a-5p | |||||||||||||||||||||||||||||||||
Wilms’ tumor | SALL4 | Long non-coding RNA (lncRNA) small nucleolar RNA host gene 12 (SNHG12) promotes proliferation, migration, and invasion and inhibits apoptosis in breast cancer by upregulating SALL4 expression via sponging miR-15a-5p; SALL4 is a direct target of miR-15a-5p | SALL2 | High | qRT-PCR, Western blot, and Luciferase reporter assay | Undescribed | SALL2 was identified as one of the 27 signature genes highly expressed by comparing tumor samples with normal fetal kidneys |
[107] |
[114] |
||||||||||||||||||||||||||||
Oncogene | [ | ] |
[132] |
Renal cell carcinoma | miR-942 | SALL1 | |||||||||||||||||||||||||||||||
Kidney | SALL3 | miR-942 affects the survival of patients with renal cell carcinoma by negatively regulating the expression of SALL1 | RNA-seq and qRT-PCR |
[108] |
[115] |
||||||||||||||||||||||||||||||||
Low | Methylation | SALL3 downregulation may contribute to genome hypermethylation similar to VHL | Tumor suppressor |
[126] |
[133] |
Prostate cancer | miR-4286 | ||||||||||||||||||||||||||||||
Wilms’ tumor | SALL4 | SALL1 | High | miR-4286 regulates proliferation and apoptosis in PCa cells by directly targeting the 3′UTR of SALL1 mRNA | qRT-PCR and Luciferase reporter assay | Undescribed | [109] |
[116] |
|||||||||||||||||||||||||||||
Undescribed | Oncogene |
[127] |
[134] |
Lung cancer | HOXA11-AS and miR-3619-5p | SALL4 | lncRNA homeobox A11 antisense (HOXA11-AS) promotes proliferation, migration, invasion, and glycolysis in non-small cell lung cancer (NSCLC) cells by upregulating SALL4 expression via sponging miR-3619-5p; SALL4 is a direct target of miR-3619-5p | qRT-PCR, Western blot, and Luciferase reporter assay |
[110] |
[117] |
|||||||||||||||||||||||||||
Osteosarcoma | ZEB2-AS1 and miR-107 | SALL4 | lncRNA ZEB2-AS1 (ZEB2-AS1) promotes proliferation, invasion, and metastasis and inhibits apoptosis in osteosarcoma cells by upregulating SALL4 expression via sponging miR-107; SALL4 is a direct target of miR-107 | qRT-PCR, Luciferase assay, and RNA pull-down assay |
[111] |
[118] |
|||||||||||||||||||||||||||||||
Hepatocellular carcinoma | miR-296-5p | SALL4 | miR-296-5p inhibits stemness potency of hepatocellular carcinoma (HCC) cells via the Brg1/Sall4 axis; Brg1 binds to the SALL4 promoter | qRT-PCR, Western blot, Luciferase reporter assay, and Chromatin immunoprecipitation (ChIP) assay |
[112] |
[119] |
|||||||||||||||||||||||||||||||
Hepatocellular carcinoma | miR-15a | SALL4 | Exosomal miR-15a reduces proliferation, migration, invasion, and survival by directly targeting SALL4 | qRT-PCR, Western blot, and Luciferase reporter assay |
[113] |
[120] |
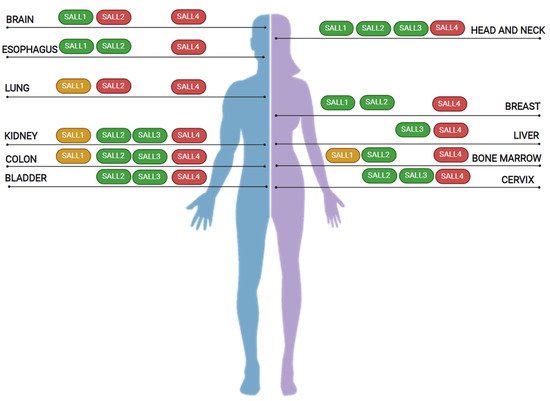
5. SALL Proteins in Cancer
Cancer Type | SALL Member | Expression Levels | Genetic Alteration/Regulation | Association With Cancer/Biological Process | Proposed Cancer Role | Ref. | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Lung | SALL1 | High |