Neutrophils and platelets exhibit a diverse repertoire of functions in thromboinflammatory conditions such as stroke. Neutrophils can enable, as well as resolve, cerebrovascular inflammation via many effector functions including neutrophil extracellular traps, serine proteases and reactive oxygen species, and pro-resolving endogenous molecules such as Annexin A1. Like neutrophils, platelets also engage in pro- as well as anti-inflammatory roles in regulating cerebrovascular inflammation. These anucleated cells are at the core of stroke pathogenesis and can trigger an ischemic event via adherence to the hypoxic cerebral endothelial cells culminating in aggregation and clot formation.
- neutrophils
- platelets
- stroke
- annexin A1
- resolution
- thromboinflammation
1. Introduction
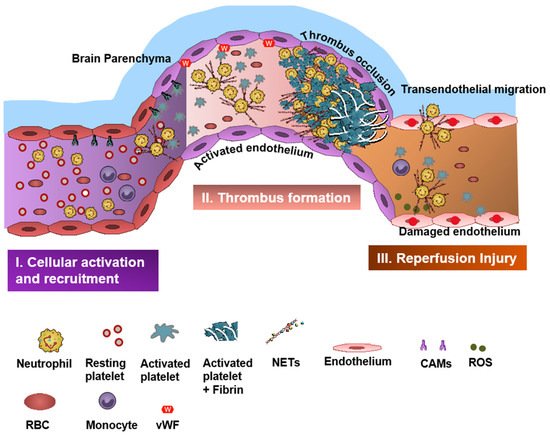
Neutrophils and platelets are key players in ischemic brain injury and its resolution [1][2][3][4][4,5,6,7]. Resolution is the physiological ability of the body to achieve homeostasis after infection or inflammation. However, in chronic inflammation, where there is an excessive and persistent inflammatory response, the process of resolution is hampered [5][6][8,9]. Acute cerebral ischemia induces a strong immune response resulting in recruitment of several subsets of leukocytes (mainly neutrophils), activation of platelets, and coagulation cascade and upregulation of cell adhesion molecules and cytokines [7][10]. Neutrophils and platelets are known for their ability to produce proinflammatory/prothrombotic mediators, thereby forming an important link between inflammation and thrombosis, a phenomenon referred to as “thromboinflammation” [1][8][9][4,11,12].
2. Neutrophils in Stroke
3. Neutrophil Serine Proteases and Thromboinflammation
Amongst Neutrophil granule serine proteases (NSPs), cathepsin G (CatG) and neutrophil elastase (NE) are particularly known to have thromboinflammatory phenotypes in various inflammatory pathologies [26][27][28][29][30][31,32,33,34,35]. NSPs can initiate and promote thromboinflammation in stroke by interacting with platelets and coagulation factors [8][11] and binding with formyl peptide receptors (FPRs) on neutrophils and platelets [27][31][32,36].4. Neutrophil-Dependent Oxidative Stress and IS
Neutrophils are rich sources of reactive oxygen species (ROS) and can contribute to harmful oxidative stress, which can further accelerate thromboinflammation. ROS production in the peri-infarct area has a major role in the pathogenesis of ischemic- and reperfusion-related brain injury [32][33][47,48]. ROS regulates neutrophil recruitment during inflammation by mainly inducing expression of adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1), and can facilitate the opening of intercellular passageways to help neutrophils transmigrate to the inflammatory tissue [34][49]. There are multiple studies that have shown that targeting ROS production may attenuate oxidative stress and inflammation, reduce edema, and help to maintain the function and integrity of the BBB [35][50]. Remote ischemic conditioning and hypothermia can also attenuate oxidant stress-induced inflammation, and non-pharmacologic adjunctive ROS-targeting therapies are currently being tested to augment neurovascular protection in IS [36][37][51,52]. ROS can also enhance thromboinflammation by inhibiting the tissue factor pathway inhibitor (TFPI), which is the only physiologic inhibitor of TF activity [38][53].5. Platelets in Stroke
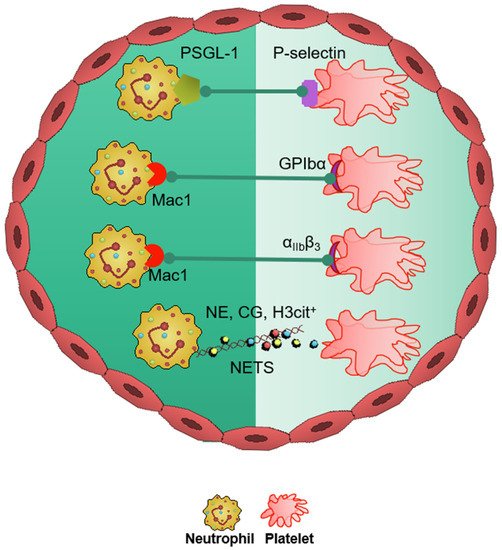
In the neurovasculature, there are distinct mechanisms of platelet-mediated thromboinflammation, which involves interaction with the neutrophils, endothelial cells, plasmatic coagulation factors, and the complement system [39][40][63,64]. In stroke, platelets and neutrophils are the first immunomodulatory cells recruited to the affected cerebral vessel where they initiate aggregation and thrombus formation [39][63]. The interaction of the platelets with the surrounding milieu, including circulating neutrophils, plays a significant role in regulating thromboinflammation [4][9][40][7,12,64]. Platelets express P-selectin on activation, which interacts with PSGL-1 to enhance neutrophil activation and recruitment at the inflammatory site. The CD40 ligand (CD40L) is found on platelets and is released on activation in the soluble circulating form, thus inducing endothelial cells to secrete chemokines and express adhesion molecules, thereby initiating a vascular inflammatory response. CD40L is also a key regulator of NPA formation and can accelerate early stages of atherosclerosis and plaque development, promote progression toward advanced atherosclerosis; and influence regulatory T cell recruitment in atherosclerosis, which is one of the main underlying causes of stroke pathogenesis [41][65]. Platelet PF4-dependent HIT can result in NPA formation and the development of thrombi enabling the pathogenesis of stroke [42][43]. Damage-associated molecular pattern molecule high-mobility group box 1 (HMGB1) is upregulated by activated platelets in multiple inflammatory diseases and has also been shown to be a critical mediator of thrombosis by regulating platelet activation, granular secretion, adhesion, and spreading [43][66]. HMGB1 effects on platelets seems to be mediated via platelet toll-like receptor 4 (TLR4) followed by MyD88/GC complex formation and activation of the cGMP-dependent protein kinase I (cGKI) [43][66]. Interestingly, platelet TLR4 also activates NET production, which can further enable stroke pathogenesis [44][67]. Platelet activation and aggregation resulting in thrombosis is further influenced by the high shear forces generated from the blood flow around the thrombus microenvironment [45][68]. The von Willebrand factor (vWF) is a key participant in the platelet-dependent thromboinflammation and stroke development [46][69]. Shear stress activates and brings conformational change to vWF, which then associates with platelet GPIbα (a subunit of GPIb-IX-V complex). This vWF–GPIbα interaction is crucial for initial platelet adhesion, which in turn facilitates platelet aggregation and adhesion in thrombotic events [47][48][70,71].6. Therapeutics in Thromboinflammation
In pre-clinical studies, engagement of the AnxA1-FPR2/ALX pathway in neutrophils as well as platelets produced significant results of mitigation and rescue of the adverse thromboinflammatory phenotype in cerebral microvessels, theoretically preventing the onset of IS as well as management of secondary I/RI-related inflammation (Figure 3) [1][3][4][49][50][4,6,7,75,82].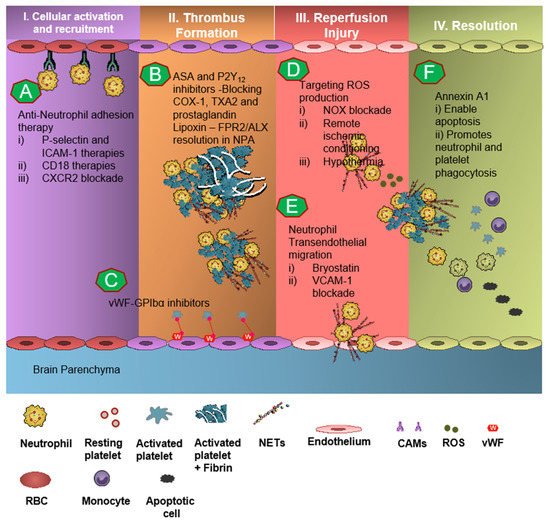
7. Targeting Neutrophil-Dependent Thromboinflammation
Neutrophil recruitment to the ischemic site and adhesion to brain endothelial cells is enabled by P-selectin and ICAM-1 [51][52][53][83,84,85]. The anti-neutrophil adhesion strategy targeting P-selectin and ICAM-1 was proven to diminish neutrophil recruitment and transmigration at the site of cerebral I/R, thereby resulting in attenuation of thromboinflammation [52][53][84,85]. CD18 (leukocyte counter-ligand to endothelial intracellular adhesion molecule-1) knockout mice conferred cerebrovascular protection in a murine model of IS, but not to CD18-deficient animals with permanent middle cerebral artery occlusion, suggesting anti-neutrophil adhesion strategies should be further tested for the management of stroke [52][84]. However, Enlimomab, a murine ICAM-1 antibody that is known to reduce leukocyte adhesion and infarct size in experimental stroke studies, was not effective in earlier clinical trials, with more adverse events such as infections and fever compared to the placebo [54][86]. Studies targeting anti-E-selectin, anti-L-selectin, and chemokine receptors had no response to minimal response in animal models of experimental IS [55][13].