Alzheimer’s disease (AD) is one of the looming health crises of the near future. Increasing lifespans and better medical treatment for other conditions mean that the prevalence of this disease is expected to triple by 2050. The impact of AD includes both the large toll on individuals and their families as well as a large financial cost to society. So far, we have no way to prevent, slow, or cure the disease. Current medications can only alleviate some of the symptoms temporarily. Many animal models of AD have been created, with the first transgenic mouse model in 1995. Mouse models have been beset by challenges, and no mouse model fully captures the symptomatology of AD without multiple genetic mutations and/or transgenes, some of which have never been implicated in human AD. Over 25 years later, many mouse models have been given an AD-like disease and then ‘cured’ in the lab, only for the treatments to fail in clinical trials.
1. Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disease, behaviourally characterised by memory loss and cognitive decline, generally in later life, which is ultimately fatal
[1]. The prevalence of AD is rapidly increasing due to an ageing population worldwide, and expected to triple between the years 2000 and 2050
[2][3][2,3]. Besides those affected, AD places a severe burden on families, carers, and the economy
[4][5][4,5]. Alois Alzheimer discovered the neuropathological hallmarks of AD in 1906
[6]. Despite many decades of research since the 1900s, a cure has remained elusive with current therapies only offering temporary symptomatic relief.
Classically, AD is characterised by plaques and tangles, both of which contain insoluble protein deposits that progressively accumulate in the brain
[7][8][9][7,8,9]. These pathological features develop over decades, and considerable effort has been devoted to their replication in short lived models. Implicit in these modelling efforts is that the relatively rare dominant genetic forms of AD represent the condition as a whole, and that the accelerated processes artificially engineered into these models accurately represents the mechanisms of a slow disease process in humans. Most models have been constructed to recapitulate the end stage pathological features, assuming that they represent the cause of the condition rather than the consequence. To a large extent, this pathology attainment strategy for AD animal model construction has driven the preclinical selection of compounds going through to human clinical trials. Well over 200 compounds have now failed to prevent, slow, or cure the disease, despite most being effective at ‘curing’ mouse models of AD
[10][11][12][10,11,12].
2. Modelling AD in Animals
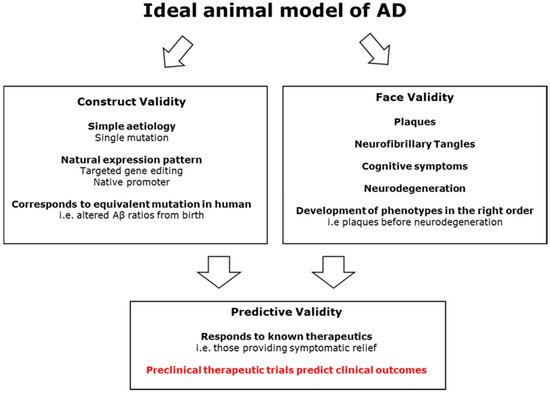
One of the most effective ways of investigating the pathogenic process of a disease is via animal models. Animal models can also be used for biomarker discovery, which can allow for early detection of disease, and for screening and safety tolerance testing of therapeutic agents. There are three main aspects of animal modelling that need to be considered: the resulting face, construct, and predictive validities
[13][40]. These relate to how well the model replicates symptoms, the biological causes, and responds to clinically effective therapeutics, respectively.
The earliest animal models of AD were created by disrupting the cholinergic system in various mammalian species using surgical methods, neurotoxins, immunotoxins, or pharmacological methods. The species targeted included mice and rats
[14][41], rabbits
[15][42], and monkeys such as the marmoset and crab eating macaque
[16][17][43,44]. The cholinergic system in the basal forebrain degenerates early in the course of AD
[18][19][45,46]. These models replicated some of the symptoms of AD such as memory impairments, and were helpful for testing the efficacy of cholinesterase inhibitors, which can offer some symptomatic relief early in the course of AD
[14][41]. These models, of course, did not develop plaques or tangles, nor did they represent the progression of the complex biochemical and cellular-level changes in AD
[20][47].
The rapid development of genetic technology and engineering from the 1980s to the present has enabled the construction of animal models that can theoretically recapitulate diseases from their underlying causes, thus increasing the construct validity of the model.
3. Small Animal Models of AD
3.1. Mouse Models
3.1.1. Plaque Pathology in Mouse Models
As a mammalian model system, mice have the advantages of a short lifespan and rapid reproduction, which facilitates timely completion of experimental protocols. They are also comparatively easy to maintain and breed in a laboratory environment. Numerous tools, data, and standardised behavioural tests have been established for assessing phenotypes in mice. The development of embryonal stem cells and targeted mutagenesis has enabled the production of models that more accurately recapitulate the aetiology of human disease state. These factors combined has resulted in mice being the most common animal models of AD.
There have been a large number of mouse models constructed in various ways, far too many to include here. We have selected a representative group of models that were either notable because they were novel at the time or have been widely used in the field.
Table 1 lists these selected mouse models.
Table 1. Selected key mouse models of AD and their major phenotypes.
| Name |
Type of
Modification |
FAD Mutations |
MAPT
Mutations |
Plaques |
Tangles |
Neurodegeneration |
Reference |
| PDAPP |
Transgenesis |
Indiana in APP |
|
X |
|
|
[21][48] |
| Tg2576 |
Transgenesis |
Swedish in APP |
|
X |
|
|
[22][49] |
| TgCRND8 |
Transgenesis |
Swedish and Indiana in APP |
|
X |
|
|
[23][50] |
| PSAPP |
Transgenesis |
Swedish in APP, M146L in PSEN1 |
|
X |
|
|
[24][51] |
| BRI-Aβ40 |
Transgenesis |
Aβ1–40 peptide |
|
|
|
|
[25][52] |
| BRI-Aβ42 |
Transgenesis |
Aβ1–42 peptide |
|
X |
|
|
[25][52] |
| 5XFAD |
Transgenesis |
Swedish, Florida, London in APP. M146L and L286V in PSEN1 |
|
X |
|
X |
[26][53] |
| JNPL3 |
Transgenesis |
|
P301L in MAPT |
|
X |
X |
[27][54] |
| rTg4510 |
Transgenesis |
|
P301L in MAPT |
|
X |
X |
[28][55] |
| 3xTg |
Transgenesis |
Swedish in APP, M146L in PSEN1 |
P301L in MAPT |
X |
X |
X |
[29][56] |
| TAPP |
Transgenesis |
Swedish in APP |
P301L in MAPT |
X |
X |
X |
[30][57] |
Plaques and tangles are the two main pathological hallmarks of AD, followed by neurodegeneration. In order to create models with high face validity, these phenotypes have been highly sought after. The first reported mouse models that developed plaque pathology were created via transgenesis (TG). Researchers introduced the human
APP gene (h
APP) containing mutations known to cause FAD. The first mouse model, the PDAPP line created in 1995, overexpressed the V717F Indiana mutation h
APP with the Platelet-Derived Growth Factor (PDGF) promoter via a minigene construct. Around 40 copies of the transgene were randomly inserted in this line at a single site, and all three major splice variants of h
APP (695, 751, and 770) were expressed. These mice developed both dense and diffuse plaque pathology by eight months of age in the entorhinal cortex, cingulate cortex, and hippocampus. By 18 months, the amyloid burden in these brain regions was thought to be greater than that seen in end stage human disease. This model also showed signs of synaptic loss, microgliosis, and astrocytosis, but no tau/tangle pathology or neurodegeneration
[21][31][48,58].
The next, and still commonly used mouse model, was the Tg2576 line, which overexpressed the K670M/N671L Swedish mutation in a transgene containing the 695 isoform of human h
APP transgene driven by the Prion Protein (PrP) promoter. Tg2576 mice develop plaques and memory deficits in a progressive manner. Similar to the PDAPP mice, they do not show the tangles or neurodegeneration
[31][32][58,59]. These mouse models developed memory deficits and synaptic loss preceding the accumulation of insoluble plaques, providing evidence for the hypothesis that it is the smaller soluble forms of Aβ that cause these symptoms
[33][34][60,61]. Several further mouse lines were also created by introducing the h
APP gene with various FAD causing mutations; most exhibited plaques and memory deficits in an age-dependant manner as well as some level of synaptotoxicity (reviewed in
[35][62]).
Some of these mouse lines were subsequently crossed to produce mouse lines with multiple
APP transgenes; the result was usually a similar phenotype that appeared at an earlier age, which shows that these mutations have cumulative phenotypic effects. One example is the TgCRND8 line, engineered with a single transgene to contain the h
APP isoform 695 with both the Swedish and Indiana mutations under the control of the Prp promoter. These mice develop plaque pathology by three months of age, with earlier signs of cognitive impairment relative to the models with a transgene carrying a single AD mutation. The brain concentration of Aβ
1–42 in this compound model at six months was equivalent to the original PDAPP mouse line at 16 months. This compound model also showed an increase in Aβ
1–42 to Aβ
1–40 ratio (now considered to be an important indication of amyloidogenesis)
[23][50]. However, these mice still did not exhibit the other major neuropathological hallmarks of AD such as the tangles and neurodegeneration.
Some of the APP overexpression mouse lines were later crossed with mice carrying a human
PSEN1 transgene (h
PSEN1) with various mutations responsible for FAD. Interestingly, mice overexpressing h
PSEN1 mutations do not develop plaques or other symptoms, but do exhibit an increased ratio of the more amyloidogenic Aβ
1–42 relative to Aβ
1–40 in the brain
[36][37][38][63,64,65]. Crossing transgenic mice that overexpressed
APP with
PSEN1 transgenic mice greatly increased amyloid pathology. An example is the crossing of Tg2576 mice (
APP Swedish mutation) with both the PS-1 line (
PSEN1 M146L mutation)
[24][38][51,65] and the
PSEN1 A246E line
[36][39][63,66]. Taken together, this animal model work helped confirm that APP metabolism, and in particular, the production of the Aβ
1–42 peptide, was affected by mutations in
APP and
PSEN1, and that these mutations are likely acting on a single pathway. This work also provided supporting evidence for the hypothesis that the Aβ
1–42 fragment is more toxic than Aβ
1–40.
Attempts to confirm the role of individual Aβ peptides led to the creation of transgenic mouse lines that selectively expressed either the Aβ
1–40 or Aβ
1–42 amyloid fragment in the absence of the h
APP transgene (BRI-Aβ40 and BRI-Aβ42)
[25][52]. These models showed that high expression of Aβ
1–40 caused no overt plaque pathology, but even low expression levels of Aβ
1–42 was sufficient to cause plaque formation in both parenchymal brain tissue and blood vessels (cerebral amyloid angiopathy).
Attempts to capture a more complete AD phenotype led to crossing transgenic mice or creating constructs to overexpress multiple transgenes and mutations within these genes. Cell loss and neurodegeneration was ultimately achieved in the 5XFAD mouse model that expressed three
APP (Swedish K670M/N671L, Florida I716V, and London V717I) and two
PSEN1 (M146L and L286V) mutations under the murine Thy-1 promoter
[26][40][53,67]. The severe phenotype again supported the hypothesis that FAD mutations have an additive effect. However tangles, which are the other main hallmark of AD, were absent in these mice.
3.1.2. Replicating AD Tau Pathology
Interestingly, unlike other mammalian species (see below), wild type mice do not develop tangles as they age
[41][68]. Mutations in the human
MAPT gene (microtubule associated protein tau), which codes for the human TAU (hTAU) protein, cause frontotemporal dementia (FTD), but not AD
[42][69]. However tangle pathology, neurodegeneration, and memory loss were seen in transgenic mice models expressing human
MAPT (h
MAPT) with FTD causing mutations. The first mouse model with this phenotype was the JNPL3 line, which expressed the 4R0N isoform of h
MAPT with the P301L mutation
[27][54]. Subsequently, a hTAU expression tetracycline repressible mouse line (rTg4510) demonstrated that the smaller soluble forms of oligomeric TAU caused memory loss and neurodegeneration
[43][44][70,71]. Many overexpression h
MAPT transgenic lines have been produced and some have been crossed with transgenic mouse lines overexpressing FAD mutations in
APP and/or
PSEN1. The resulting lines demonstrated that the mechanisms leading to amyloid and TAU pathology interact. The 3xTg mice (Swedish mutation in
APP, M146V in
PSEN1, and P301L in
MAPT) develop plaques before tangles
[44][45][71,72], as observed in AD patients. A line developed by crossing the aforementioned
APP mutant mice Tg2576 with the
MAPT JNPL3 mice (called the TAPP line) altered the spatial distribution of tangles in the brain relative to original TAU expressing strain, with TAPP mice exhibiting tangles in the subiculum, hippocampus, and isocortex that were not present in JNPL3 mice. TAPP mice also had greatly increased numbers of tangles in the olfactory cortex, entorhinal cortex, and amygdala. This suggests that Aβ fibril deposition can alter the amount and distribution of insoluble TAU as tangles
[30][57].
3.1.3. Construct Validity of Transgenic Mouse Models of AD
Although mice expressing a transgene with a single FAD mutation display some symptoms of the disease, it is evident from the literature that three or more AD and FTD associated mutations are required to replicate the majority of the human pathology. In contrast, multiple mutations have not been reported in humans with AD, and in nearly all cases of FAD, only a single mutation is required to develop the entire phenotype.
There are good reasons for the requirement of a compound approach to create equivalent AD pathology. Unlike human h
APP, the proteolytic cleavage products of murine
App (m
App) do not naturally form plaques. This is due to three amino acid substitutions in the amyloid beta sequence compared to human (
Figure 12), which reduces the ability of murine Aβ peptides to aggregate
[45][72]. In addition, murine β-secretase enzymes typically cleave m
App to form Aβ
11-x, even though it cleaves h
APP to form Aβ
1-x [46][47][73,74]. Deposition of cleavage products from m
App is only apparent in models with high expression levels of m
App and only after an extended period. This is one of the reasons why h
APP is typically used instead
[48][75].
Figure 12. A comparison of the human and mouse Aβ peptide sequence, showing the three amino acid substitutions responsible for the functional difference between the two.
The ratio of h
APP isoforms differs within brain regions and also in other organs. The ratio also changes during the course of development and ageing
[49][50][76,77]. The two longer isoforms of h
APP, 751 and 770, are more prevalent in the AD brain relative to healthy controls
[51][78]. Overexpressing the h
APP 751 isoform also causes more obvious amyloid pathology in mice than overexpressing the short (APP695) isoform
[52][79]. The pathology generated in a mouse model therefore depends on which of the three isoforms of h
APP is overexpressed, or whether the full h
APP gene sequence is used.
Several different promoters have been used to drive overexpression of h
APP in mouse models of AD including the promoters for PDGF-B (platelet derived growth factor B-chain) and the PrP (prion protein gene motifs). Different promoters drive different levels and spatial patterns of expression including outside the brain. For example, the PDGF-B and Thy-1 (thymocyte differentiation antigen 1) promoters are neuron-specific
[53][54][80,81], while the PrP promoter has less specificity, also driving expression in glial cells and other non-brain tissue
[55][82]. The Thy-1 promoter included in the construct to make the APP23 model (Swedish mutation in
APP) is active only after birth, preventing potential developmental effects
[56][83]. Various Tet-controlled lines have been created that allow for more control over the timing and location of transgene expression, but have the added complication of requiring an extra transgene
[57][58][59][84,85,86]. All of these promoters are selected for ease of use or particular benefits, but because none of them are the endogenous promoter, the natural expression pattern of
APP is not replicated in any of the models.
3.1.4. Murine APP Knock in Models
In an attempt to overcome the limitations of
APP TG models, a small number of knock in (KI)
App models have been created with targeted gene editing. Inserting selected mutations in the endogenous genes should mean expression is quantitatively, spatially, and temporally appropriate. Mouse
App was ‘humanised’ in these models by converting the codons for the three amino acids that differ between human and mice in the Aβ coding portion of m
App. This allows murine BACE1 to cleave mAPP at the human equivalent position
[60][61][62][63][87,88,89,90]. These mice did not develop overt phenotypes such as memory deficits, synaptic loss, and/or plaque pathology. These phenotypes only became evident after the insertion of multiple
APP mutations (combinations of Swedish, London, Dutch, Iberian, and Artic)
[64][91], and usually only after breeding to homozygosity in concert with homozygous FAD
PSEN1 mutations
[63][65][90,92].
The necessity of including multiple mutations to induce human equivalent disease confounds the use of these models, but they have helped differentiate between phenotypes due to the TG process, and those that represent the disease in a mouse. Consistent phenotypes observed in TG and KI models include plaque formation, changes to glial cells and astrocytes, and lowered rates of hippocampal neurogenesis, although some artifacts such as transgene calpain activation have been noted
[66][67][68][93,94,95]. The presence of cognitive impairment appears to vary more between KI models than TG models. The KI models with cognitive impairment have plaque pathology prior to memory impairment, unlike the commonly used TG mice models
[62][69][89,96]. Memory impairment following plaque formation is the order of events seen in patients
[70][97], so KI models do appear to more faithfully replicate symptom clusters. Despite this, the higher variability of phenotypes in KI models, along with their milder symptom profile, means that transgenic models are still widely used.
3.1.5. Murine PSEN1 Knock in Models
Murine
Psen1 (m
Psen1) does not require ‘humanising’ like the m
App and (m
Psen1) models made with targeted gene editing by introducing FAD mutations, which show similar phenotypes to TG h
PSEN1 mouse lines
[71][72][98,99]. Whether they are created by transgenesis or targeted gene editing, in the absence of h
APP or humanised m
App, all modelled
PSEN1 mutations only increased the level of murine Aβ
1–42 in mice, had little or no effect on murine Aβ
1–40 levels, and did not result in AD equivalent symptoms
[38][73][74][75][65,100,101,102]. For this reason, more recently generated KI mouse models carrying a
PSEN1 mutation usually incorporate a transgene overexpressing mutant h
APP. The resulting animals have a more acute phenotype than
APP mutations alone
[76][77][78][103,104,105].
3.1.6. Construct Validity of MAPT Mouse Models
Compared to hTAU with six isoforms (named 4R2N, 4R1N, 4R0N, 3R2N, 3R1N, and 3R0N)
[79][80][81][106,107,108] murine TAU (mTAU) only has three of the human equivalent isoforms (4R0N, 4R1N, 4R2N). There is also variability in TAU protein conservation. Some regions of mTAU tau are very similar to hTAU, while other regions differ greatly. There are species-specific differences in the presence of different isoforms during development, and spatially across the brain
[81][82][108,109]. In TG models, the presence of endogenous mouse Mapt (m
Mapt) can alter the splicing ratios of introduced h
MAPT [83][84][110,111].
As stated above, unlike in humans, tangles do not form naturally with age in the mouse brain. Indeed, it appears that replacing the m
Mapt gene with the human equivalent, and in some lines with a FTD mutation, is necessary to create a TAU dysfunction phenotype in mice
[85][112]. The inclusion of FTD mutations to ensure a tangle phenotype in murine models is a major issue for construct validity. There are probably better models of frontotemporal dementia and other tauopathies than AD, even though they have provided insights about TAU toxicity
[29][30][56,57]. Unexpected non-disease associated deficits have been found in some models, for example, the commonly used JNPL3 line (P103L mutation in
MAPT) has motor impairments and develops eye irritations
[27][86][54,113]. Further the Tau P301S line develops severe paraparesis at 5–6 months
[87][114]. However severe motor impairment is not usually observed in AD until late in the disease course
[88][115].
3.1.7. Predictive Validity of Murine Models
Almost no mouse model of AD has shown predictive validity in human clinical trials to date, despite many therapeutic agents ‘curing’ a mouse of AD symptoms (for reviews, see
[85][86][112,113]). Those that have been successful were based on the cholinergic system or NMDA receptors and only provide temporary symptomatic relief. While symptomatic relief is important, the predicted increase in the prevalence of AD means that finding a method to prevent or cure the disease is now becoming an urgent priority.
In addition to drug failures, there is the issue of differences in drug metabolism between species; something well tolerated in mice may not be so in humans
[89][90][116,117]. Many clinical trials have failed to make it to later stages due to adverse side effects, which were not present in mice. For example, immunisation of mice with Aβ
1–42 (named AN1792 in the clinical trial) was able to lower the volume of plaque material in the brain and preserve cognitive function. Unfortunately, this approach failed to show benefits in clinical trials and 6% of the immunised patients developed meningoencephalitis
[91][92][118,119]. The adverse effects were thought to be due to a T-cell response in humans against the large Aβ
1–42 fragment. Subsequent immunisation trials with smaller epitopes that were beneficial in mice including the drugs Bapineuzumab
[93][120] and Solanezumab
[94][121] showed a similar lack of efficacy and/or adverse side effects
[95][96][97][122,123,124].
To date, well over 200 compounds have failed to affect the disease course
[10], and this appears to have led to some controversial decisions. Recently, the drug aducanumab (sold as Aduhelm) was approved by the FDA through an accelerated approval pathway, on the condition that follow-up trials are performed to determine efficacy. This drug showed mixed results in clinical trials, with a benefit seen at the highest dose, but only in one of the two trials. Given that 35% of patients developed brain swelling (cerebral adema) and 19% brain bleeds (intracerebral haemorrhage), there are serious safety considerations
[98][125]. It is clear that models of AD with higher predictive validity are desperately needed.
3.1.8. Murine Model Summary
In summary, while successive generations of mouse models come closer to attaining the desired symptom clusters, this has created a trade-off between face and construct validity. The drive to replicated AD’s defining features of both plaques and tangles in a model is understandable. However, the inclusion of multiple mutations, with some from a different condition altogether, brings the construct validity of these models into question. Do they represent the disease process or a derived phenocopy? Discovering the mechanism by which amyloid pathology triggers TAU dysfunction would be invaluable for understanding the disease. Unfortunately, it appears that the mouse is too genetically and physiologically dissimilar to be able to capture this transition, even with genetic modification. The lack of translatability of treatments developed using these models is suggestive that they may not adequately represent AD. Work to understand the mechanistic nature of various FAD and MAPT mutations continues, and many of the aforementioned models are still utilized. Mouse models of LOAD variants have also been made including APOE and TREM2
[99][100][101][126,127,128]. It is likely that mechanistic work via mouse models will continue as more LOAD disease related genetic or environmental risk factors are discovered. However, over the last ten years, there have been repeated calls for new models of AD that can show predictive validity, causing researchers to look outside mice.
Figure 23 summarises the desired qualities of an AD model, showing how improved construct validity could lead to higher translatability in clinical trials.
Figure 23. A diagram showing how a model of AD with high face and construct validity is likely to improve predictive validity and lead to effective therapies.
3.2. Rat Models of AD
Rats are an attractive model system because they are genetically and physiologically more similar to humans than mice. They display more complex behaviour, and numerous assessment methods have been developed for mood and cognition for this species
[20][102][103][104][47,129,130,131]. The first rat models of AD were based on knowledge from mouse models, designed to express h
APP with FAD mutations such as the UKUR25 line (with the Swedish and Indiana
APP mutations, with the M146L
PSEN1 mutation
[105][132]) or the McGill-R-Thy1-APP line (with the Swedish and Indiana h
APP mutations, expressed under the murine Thy1.2 promoter
[106][107][133,134]). Interestingly, these models failed to develop the plaques seen in mice, but did accumulate intracellular Aβ and developed memory deficits seen in the equivalent mouse models. Rat models that exhibited plaque pathology were finally created in the mid 2000s, sometimes with differing phenotypes from their murine genetic equivalents. The rat TgF344-AD line carries both Swedish h
APP and
PSEN1 ΔE9 mutations, driven by the same murine PrP promoter
[108][135]. This line develops both plaque and tangle-like pathology with loss of neurons. Interestingly the tangle-like structures appear despite the non-inclusion of a h
MAPT transgene, even though tangles are not naturally seen in aged rats. This may be because unlike m
Tau, rat
Tau (r
Tau) is spliced to create all six human equivalent isoforms
[109][110][136,137]. Rat models expressing h
MAPT with FTD mutations have also been developed, some of which exhibit tangles, while all show increased phospho-TAU in the brain and develop cognitive symptoms
[111][112][113][138,139,140]. The development of Tau pathology, along with their larger brain and more complex behaviours, may confer on these models the potential to improve our understanding of AD.
4. Large Animal Models in AD Research
In pursuit of more translatable results across the medical sciences, researchers are more frequently turning to large animal models, particularly those that are evolutionarily closer to humans and have longer lifespans, thus making them better suited to recapitulate complex human diseases, especially late onset disorders
[114][115][116][141,142,143]. Massively overexpressing transgenes speeds up the development of a phenotype, but also leads to acute inflammatory processes not present in human AD. Small mammals also have a smooth (lissencephalic) brain, while most larger mammals including humans have a more complex convoluted (gyrencephalic) brain. This makes larger mammals ideal for studying neurological disorders.
Many large animals naturally develop plaques and/or tangles as they age, and these features may be the norm in larger animals. They have been found in many primate species, and across a range of large herbivorous and carnivorous animals (summarised in
Table 2). This propensity to develop plaques appears to be due, at least in part, to conservation of the amyloid beta peptide sequence in most mammals
[117][144]. Of note is that in some large animals (e.g., dogs, sheep), only one of either plaques or tangles were originally identified, but later research revealed both
[118][119][145,146]. They are typically found in aged animals, and vary widely in density between individuals of a species, so it is entirely possible that both hallmarks of AD will eventually be found in most large animals. Age related neurodegeneration has not been extensively studied in most species (
Table 2), however, many are known to develop cognitive decline with age. Cognitive decline has rarely been studied in detail outside of humans, dogs, cats, and some primates.
Table 2. A summary of the large animals in which plaque, tangle pathology, or neurodegeneration with older age has been identified.
| Species |
Scientific Name |
Plaques |
Tangles |
Neurodegeneration |
References |
| Chimpanzee |
Pan troglodytes |
X |
X |
|
[120][121][147,148] |
| Orang-Utan |
Pongo spp. |
X |
|
|
[122][149] |
| Western Gorilla |
Gorilla |
X |
X |
|
[123][124][150,151] |
| Eastern Gorilla |
Gorilla beringei |
X |
X |
|
[125][152] |
| Cynomolgus Monkey |
Macaca fascicularis |
X |
X |
|
[126][127][128][129][153,154,155,156] |
| Rhesus Macaque |
Macaca mulattas |
X |
X |
|
[130][131][157,158] |
| Stump Tailed macaque |
Macaca arctoides |
X |
X |
|
[132][159] |
| Vervet Monkey |
Chlorocebus aethiops |
X |
X |
|
[133][160] |
| Baboon |
Papio hamadryas |
X |
X |
|
[134][135][136][161,162,163] |
| Cotton Topped Tamarin |
Saguinus oedipus |
X |
|
|
[137][164] |
| Mouse Lemur |
Microcebus murinus |
X |
X |
X |
[138][139][140][165,166,167] |
| Common Marmoset |
Callithrix jacchus |
X |
X |
|
[141][142][168,169] |
| Squirrel Monkey |
Saimiri sciureus |
X |
|
|
[143][144][170,171] |
| Pigs |
Sus domesticus |
X * |
X * |
|
[145][172] |
| Domestic Sheep |
Ovis aries |
X |
X |
|
[118][146][147][148][145,173,174,175] |
| Domestic Goat |
Capra hircus |
|
X |
|
[146][173] |
| Bactrian Camel |
Camelus bactrianus |
X |
|
|
[149][176] |
| Reindeer |
Rangifer tarandus |
|
X |
|
[150][177] |
| American Bison |
Bison |
|
X |
|
[150][177] |
| Domestic Dog |
Canis familiaris |
X |
X |
X |
[151][152][153][154][155][178[,179156,180,181],182,183] |
| Domestic Cat |
Felis catus |
X |
X |
X |
[157][158][184,185] |
| Leopard Cat |
Prionailurus bengalensis |
X |
X |
|
[159][186] |
| Polar Bear |
Ursus maritimus |
X |
|
|
[160][187] |
| Brown Bear |
Ursus arctos |
|
X |
|
[160][187] |
| Black Bear |
Ursus americanus |
X |
|
|
[161][188] |
| Wolverine |
Gulo |
X |
X |
|
[162][189] |
| Harbor Seal species |
Phoca largha, Phoca vitulina |
X |
X |
|
[163][190] |
| Sea Lion species |
Eumetopias jubatus, Zalophus californianus, Neophoca cinerea |
X |
X |
|
[163][190] |
| Walrus |
Odobenus rosmarus |
X |
X |
|
[163][190] |