Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Motoi Kanagawa and Version 2 by Lindsay Dong.
Dystroglycanopathy is a collective term referring to muscular dystrophies with abnormal glycosylation of dystroglycan. At least 18 causative genes of dystroglycanopathy have been identified, and its clinical symptoms are diverse, ranging from severe congenital to adult-onset limb-girdle types.
- muscular dystrophy
- glycosylation
- dystroglycan
1. Introduction
Muscular dystrophy is a group of genetic disorders with progressive muscle weakness, and >50 causative genes have been identified (www.musclegenetable.fr, accessed on 1 November 2021). Dystroglycanopathy (DGpathy) is a collective term referring to muscular dystrophies with abnormal glycosylation of dystroglycan (DG). DG was first identified as a glycoprotein that interacts with dystrophin, the causative gene product of Duchenne muscular dystrophy, in skeletal muscle [1]. Once translated, DG is cleaved into α and β subunits, both of which are expressed in muscle cell membranes, interacting with each other in a non-covalent fashion. αDG is an extracellular subunit containing an exceedingly large number of sugar chains, and its binding to basement membrane and synaptic proteins is mediated by the sugar chains. Its known ligands include molecules containing laminin G-domain, such as laminin and agrin [2]. On the other hand, βDG is a transmembrane protein that binds to αDG on the cell surface and to dystrophin intracellularly. Since dystrophin binds to the actin cytoskeleton, DG plays a role in connecting the basement membrane and cytoskeleton across the cell membrane.
In the early 2000s, αDG sugar chain abnormalities and loss of laminin-binding activity were reported in specimens of patients with Walker–Warburg syndrome (WWS), muscle–eye–brain disease (MEB), Fukuyama congenital muscular dystrophy (FCMD), congenital muscular dystrophy 1C, and limb-girdle muscular dystrophy (LGMD) 2I (alternatively called LGMDR9) [3][4][5][6][3,4,5,6]. Because DG and dystrophin are normally expressed in cell membranes in the tissues of these patients, αDG sugar chain abnormalities are thought to disrupt the coordination between the basement membrane and the cytoskeleton [7]. Since then, cases showing similar sugar chain abnormalities have been reported, which led to the establishment of the disease concept of DGpathy. To date, at least 18 causative genes of DGpathy have been identified [8] (Table 1).
Table 1. DGpathy genes and gene product functions.
| DGpathy Genes | Gene Functions |
|---|---|
| POMT1 | Protein O-mannosyl-transferase as a part of the POMT1/2 complex |
| POMT2 | Protein O-mannosyl-transferase as a part of the POMT1/2 complex |
| POMGNT1 | Protein O-mannose β1,2-N-acetylglucosaminyltransferase; Core M1 synthesis |
| POMGNT2 | Protein O-mannose β1,4-N-acetylglucosaminyltransferase; Core M3 synthesis |
| B3GALNT2 | β1,3-N-acetylgalactosaminyltransferase; Core M3 synthesis |
| POMK | Protein O-mannose kinase; phosphorylation of Core M3 |
| FKTN | Ribitol phosphate transferase; tandem ribitol synthesis |
| FKRP | Ribitol phosphate transferase; tandem ribitol synthesis |
| ISPD/CRPPA | CDP-ribitol pyrophosphorylase; synthesis of CDP-ribitol (donor substrate of FKTN/FKRP) |
| TMEM5/ RXYLT1 |
Ribitol-5-phosphate xylosyltransferase; synthesis of linker structure between tandem ribitol and matriglycan |
| B4GAT1 | β1,4-Glucuronyltransferase; synthesis of linker structure between tandem ribitol and matriglycan |
| LARGE | Xylosyl- and glucuronyltransferase; matriglycan synthesis |
| DAG1 | Dystroglycan |
| GMPPB | GDP-mannose pyrophosphorylase required for the formation of GDP-Man; Dolichol-phosphate-mannose synthesis |
| DPM1 | Dolichol-phosphate-mannose synthase; Dolichol-phosphate-mannose synthesis |
| DPM2 | Dolichol-phosphate-mannose synthase; Dolichol-phosphate-mannose synthesis |
| DPM3 | Dolichol-phosphate-mannose synthase; Dolichol-phosphate-mannose synthesis |
| DOLK | Dolichol kinase required for the formation of dolichol-phosphate; Dolichol-phosphate-mannose synthesis |
DGpathy exhibit a broad clinical spectrum, ranging from severe congenital muscular dystrophies, such as WWS and FCMD, to mild, adult-onset LGMD [9]. This is likely due to the effects of mutations involving the functions of gene products (enzymatic activity), rather than to variation in causative genes. In addition, one of the characteristics of DGpathy is the involvement of central nervous system disorders, such as malformation of the brain (type II lissencephaly) and mental retardation. Brain lesions in DGpathy have also shown a wide range of clinical symptoms, ranging from severe malformation of the brain to lesions with only mental retardation without structural abnormalities [10][11][10,11].
2. Sugar Chain Structure of DG and Functions of Causative Genes of DGpathy
The sugar chain structure involved in the ligand binding activity of DG and the enzymes involved in its biosynthesis are shown in Figure 1. O-Mannosyl glycosylation is required for the binding of DG to its ligands, and some of the causative genes of DGpathy encode enzymes involved in the biosynthesis of sugar chains named Core M1 (galactose-β1,4-N-acetylglucosamine-β1,2-mannose-; Gal-β1,4-GlcNAc-β1,2-Man-) and Core M3 (N-acetylgalactosamine-β1,3-N-acetylglucosamine-β1,4-mannose-; GalNAc-β1,3-GlcNAc-β1,4-Man-). The protein-O-mannose transferase (POMT) 1-POMT2 complex functions to transfer a mannose moiety to serine/threonine residues of DG [12]. Because the POMT1-POMT2 complex uses dolichol phosphate mannose (Dol-P-Man) as a donor substrate, mutations in the genes (GMPPB, DPM1/2/3, and DOLK) that encode enzymes involved in its biosynthesis have been reported to cause DGpathy (Table 1) [8]. The sugar chains of Core M1 are extended by protein O-mannose N-acetylglucosaminyltransferase (POMGNT1) [13], while those of Core M3 are extended by POMGNT2 and β1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2) [14].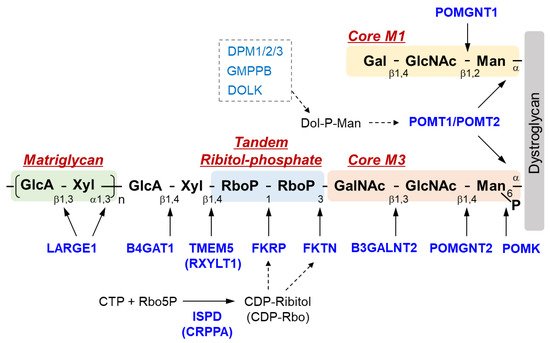
Figure 1. Overview of DG sugar chain structure and modifying enzymes. Man, mannose; GlcNAc, N-acetylglucosamine; GalNAc, N-acetylgalactosamine; RboP, ribitol phosphate; Xyl, xylose; GlcA, glucronic acid; Gal, galactose; Rbo5P, ribitol-5-phosphate; POMK, Protein O-mannose kinase; POMGNT, Protein O-mannose N-acetylglucosaminyltransferase; B3GALNT2, β1,3-N-acetylgalactosaminyltransferase 2; TMEM 5, Transmembrane protein 5; RXYLT1, ribitol-5-phosphate xylosyltransferase 1; B4GAT1, β-1,4-glucuronyltransferase 1; LARGE1, like-acetylglucosaminyltransferase 1/ LARGE xylosyl- and glucuronyltransferase 1; POMT, protein O-mannosyl-transferase; FKTN, fukutin; FKRP, Fukutin-related protein; ISPD, isoprenoid synthase domain-containing protein; CRPPA, CDP-ribitol pyrophosphorylase A.
34. Pathological Mechanism of DGpathy
3.1. Muscular Dystrophy
4.1. Muscular Dystrophy
In DGpathy, the loss of ligand binding activity of DG due to sugar chain abnormalities is thought to disrupt association with the basement membrane, which reduces the physical stability of the muscle cell membrane, making necrosis more likely to occur [7]. This pathological mechanism suggests that DGpathy patients exhibit muscle pathology similar to those of patients with dystrophin-deficient Duchenne-type muscular dystrophy (DMD). However, some DGpathy patients clearly have more severe disease manifestations than DMD patients, which cannot be explained by only the disruption of association between the basement membrane and cell membrane. Muscle fiber-selective MCK-fukutin-cKO mice present only extremely mild muscular dystrophy, suggesting that there are factors other than susceptibility of muscle fibers to necrosis [26][27][38,39]. Muscle progenitor cell-selective Myf5-fukutin-cKO mice exhibit a decreased number of muscle satellite cells and decreased proliferative activity and differentiation activity of muscle progenitor cells with progression of the pathological condition, which results in impaired muscle regeneration [27][39].
In addition, a recent study using induced pluripotent stem (iPS) cells derived from FKRP-deficient LGMD2I or WWS patients revealed decreased autophagy and increased apoptosis in myotubes, indicating that alterations in cell homeostasis may be involved in DGpathy pathogenesis [28][53].
3.2. Central Nervous System Abnormalities
4.2. Central Nervous System Abnormalities
Brain abnormalities in DGpathy patients are diverse, ranging from severe brain malformation and associated mental retardation/refractory epilepsy to average intelligence with little brain malformation [10][11][10,11]. Although an association between the type of gene mutation and the severity of the disease has been suggested, the underlying mechanism of the broad clinical spectrum of brain abnormalities in DGpathy patients remains unclear. DG is expressed in the termini of radial glia in the developing cerebral cortex, contributing to maintenance of the glia limitans-basement membrane complex. DG sugar chain abnormalities impair its binding to the basement membrane, disrupting the glia limitans-basement membrane complex [29][30][31][29,57,58]. The protrusion of neuronal cells from the site of basement membrane disruption into the subarachnoid space is thought to be the major pathological mechanism leading to type II lissencephaly [32][59].3.3. Cardiomyopathy
4.3. Cardiomyopathy
Heart failure, along with respiratory disorders, is the leading cause of death in DGpathy patients. Many FCMD patients, for example, have left ventricular systolic impairment despite the absence of left ventricular hypertrophy and fibrosis is found at autopsy [33][65]. In addition, dilated cardiomyopathy in fukutin-mutated patients with very mild muscular lesions has been reported [34][66]. As with skeletal muscle, sugar chain abnormalities are thought to cause impaired association between the basement membrane and cell membrane [35][67], but cardiomyopathy associated with DGpathy is generally mild in many cases. With regards to model mice, no myocardial lesions or cardiac dysfunction are observed in young (up to 24 weeks old) striated muscle-selective MCK-fukutin-cKO mice. However, despite the absence of cardiac hypertrophy, mice older than 1 year of age exhibit fibrosis in addition to decreased cardiac function, such as chamber dilation during diastole and decreased fractioning shortening, reproducing the cardiac pathology seen in FCMD patients [36][41]. Moreover, the contractile property of cardiomyocytes isolated from MCK-fukutin-cKO mice is reduced, and intracellular Ca2+ handling during excitation-contraction coupling is disrupted. Similarly, mild pathological changes and decreased cardiac function have been reported in FKRP-mutant mice [37][68].45. Treatment Methods
4.1. Gene Therapy
5.1. Gene Therapy
Because DGpathy is a single-gene disorder, gene therapy is considered a simple treatment strategy. As mentioned above, we showed that vulnerability of muscle fibers triggers the onset of DGpathy and that poor muscle regeneration due to decreased function of muscle progenitor cells is associated with pathological severity and progression, using skeletal muscle-selective fukutin cKO mice [27][39]. Since glycosylation status in muscle progenitor cells and myoblasts changes during differentiation [21], the expression of glycosyltransferases may also be strictly controlled. Therefore, these cells are not suitable targets for gene therapy that uses promoters constitutively inducing gene expression. On the other hand, selective gene rescue in muscle fibers is expected to suppress myonecrosis that triggers muscle regeneration and disease onset, although it does not improve poor muscle regeneration. In fact, the administration of an adeno-associated viral vector that allow a muscle fiber-selective expression of the fukutin gene dramatically ameliorates muscular dystrophy in Myf5-fukutin-cKO mice that exhibit muscle regeneration abnormalities [27][39]. The effectiveness of gene therapy for DGpathy has also been demonstrated in FKRP-mutant mice and Largemyd mice [38][39][40][69,70,71]. Dhoke et al. recently reported a gene correction approach with the CRISPR-Cas9 gene editing for FKRP-deficient DGpathy to enable cell therapy [41][72]. In this study, the authors corrected FKRP mutations in iPS cells derived from FKRP-deficient DGpathy patients using homology-directed repair to knock in the wild-type sequence, differentiated the gene-corrected cells into myogenic progenitors, and then transplanted them in the FKRP-mutant mice. Results showed restoration of glycosylation in engrafted muscles, indicating a potential of the combination of gene editing and iPS cell-derived myogenic cell transplantation as a therapeutic approach in the future.