Utilizing CO2 as a sustainable carbon source to form valuable products requires activating it by active sites on catalyst surfaces. These active sites are usually in or below the nanometer scale. Some metals and metal oxides in this scale dimension can catalyze the CO2 transformation reactions. Herein, CO2 activation on metal-based catalyst surfaces and how their structures impact the activation process are highlighted.
- CO2
- CO2 activation
- metal oxide nanoparticles
1. Introduction
2. Activation of CO2 on Heterogeneous Catalyst Surface
The interaction of CO2 molecules with metal-based surfaces has attracted intense research attention for a long time. Reduction of CO2 by H2 has been studied on some transition metals (e.g., Cu, Co, and Ni) and metal oxide catalysts (e.g., TiO2, CeO2, and In2O3). The catalytic surfaces for the activation of CO2 include metal sites, metal-oxide interfaces, and oxygen vacancies. For facile CO2 activation and conversion, efforts should be made to know, design, and optimize functional sites in heterogeneous catalysts, which involves constructing surface sites with a charge density gradient capable of reorganizing the electronic structures of CO2 and polarizing the adsorbed species [11]. Metal-based catalysts are capable of effectively activating CO2 even in the absence of hydrogen [12]. These include pure metal surfaces, doped or promoted metal surfaces, and supported metal nanoparticles. On supported metal surfaces, CO2 activation occurs mainly through acceptance of charge from the metal; the oxide support plays a big part in the activation process through different mechanisms such as acid sites interactions and provision of oxygen vacant sites [13]. The metal surface is also important in the dissociation of H2 in CO2 hydrogenation reactions [3].2.1. CO2 Activation on Representative Pure Metals
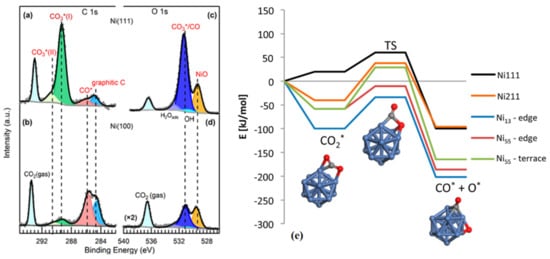
2.2. CO2 Activation on Bimetallic/Alloyed Catalyst Surfaces
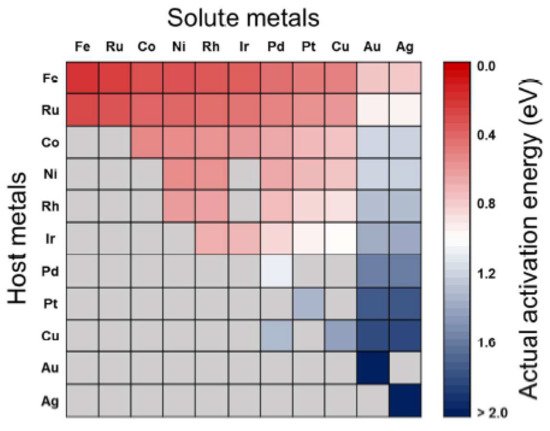
2.3. CO2 Adsorption and Activation on Metal Oxide Surfaces
2.3.1. Metal Oxide
2.3.2. Characteristic Adsorption of Representative Metal Oxides
3. Conclusion
CO2 is a kinetically stable molecule that requires high energy input for the C–O bond breaking. Its proper activation can reduce the high energy barrier substantially, easing conversion by various processes. The CO2 activation is an important step that precedes the conversion of CO2 to chemicals and fuels. It can be effected in the presence of a catalyst by altering the CO2 electronic and molecular properties. Upon accepting an extra electron from a substrate, the neutral CO2 molecule forms an anion with a full charge (CO2–) or partial charge (CO2δ–). Some metals and metal oxides are efficient catalysts for CO2 conversion reactions; thus, they should be good for CO2 activation. In general, metal nanoparticles serve as active sites for electron transfer, with certain factors such as change in morphology of metal particles, nanoparticle size, adsorption mode and configuration, and chemical ordering as the CO2 activation marker. The interaction of CO2 with some pure metals is rather weak but can be improved by incorporating promoters (e.g., alkali metals) with low electronegativity. Metal oxide nanoparticles are utilized as supports or as catalysts for CO2 conversion. Their surfaces comprise both metal (Mn+) and oxygen (O2–) ions, which can act as active sites for CO2 activation. They can activate CO2 by coordinating to one or two adjacent metal sites through the terminal oxygen atoms of the CO2 or by interaction of the carbon atom of CO2 with surface oxygen sites. A particularly interesting feature in metal oxides is the oxygen vacancies that facilitate CO2 adsorption and activation.
CO2 is a kinetically stable molecule that requires high energy input for the C–O bond breaking. Its proper activation can reduce the high energy barrier substantially, easing conversion by various processes. The CO2 activation is an important step that precedes the conversion of CO2 to chemicals and fuels. It can be effected in the presence of a catalyst by altering the CO2 electronic and molecular properties. Upon accepting an extra electron from a substrate, the neutral CO2 molecule forms an anion with a full charge (CO2–) or partial charge (CO2δ–). Some metals and metal oxides are efficient catalysts for CO2 conversion reactions; thus, they should be good for CO2 activation. In general, metal nanoparticles serve as active sites for electron transfer, with certain factors such as change in morphology of metal particles, nanoparticle size, adsorption mode and configuration, and chemical ordering as the CO2 activation marker. The interaction of CO2 with some pure metals is rather weak but can be improved by incorporating promoters (e.g., alkali metals) with low electronegativity. Metal oxide nanoparticles are utilized as supports or as catalysts for CO2 conversion. Their surfaces comprise both metal (Mn+) and oxygen (O2–) ions, which can act as active sites for CO2 activation. They can activate CO2 by coordinating to one or two adjacent metal sites through the terminal oxygen atoms of the CO2 or by interaction of the carbon atom of CO2 with surface oxygen sites. A particularly interesting feature in metal oxides is the oxygen vacancies that facilitate CO2 adsorption and activation.
References
- Lim, E.; Heo, J.; Zhang, X.; Bowen, K.H.; Lee, S.H.; Kim, S.K. Anionic Activation of CO2 via (Mn–CO2)–Complex on Magic-Numbered Anionic Coinage Metal Clusters Mn–(M= Cu, Ag, Au). J. Phy. Chem. A 2021, 125, 2243–2248.
- Alvarez-Garcia, A.; Flórez, E.; Moreno, A.; Jimenez-Orozco, C. CO2 activation on small Cu-Ni and Cu-Pd bimetallic clusters. Mol. Catal. 2020, 484, 110733.
- Álvarez, A.; Borges, M.; Corral-Pérez, J.J.; Olcina, J.G.; Hu, L.; Cornu, D.; Huang, R.; Stoian, D.; Urakawa, A. CO2 activation over catalytic surfaces. ChemPhysChem 2017, 18, 3135–3141.
- Nakamura, S.; Hatakeyama, M.; Wang, Y.; Ogata, K.; Fujii, K. A basic quantum chemical review on the activation of CO2. In Advances in CO2 Capture, Sequestration, and Conversion; ACS Publications: Washington, DC, USA, 2015; pp. 123–134.CO2. In Advances in CO2 Capture, Sequestration, and Conversion; ACS Publications: Washington, DC, USA, 2015; pp. 123–134.
- Mendes, P.C.; Verga, L.G.; Da Silva, J.L. Ab initio screening of Pt-based transition-metal nanoalloys using descriptors derived from the adsorption and activation of CO2. Phys. Chem. Chem. Phys. 2021, 23, 6029–6041.
- Koppenol, W.; Rush, J. Reduction potential of the carbon dioxide/carbon dioxide radical anion: A comparison with other C1 radicals. J. Phys. Chem. 1987, 91, 4429–4430.
- Das, S.; Pérez-Ramírez, J.; Gong, J.; Dewangan, N.; Hidajat, K.; Gates, B.C.; Kawi, S. Core–shell structured catalysts for thermocatalytic, photocatalytic, and electrocatalytic conversion of CO2. Chem. Soc. Rev. 2020, 49, 2937–3004.
- Modak, A.; Bhanja, P.; Dutta, S.; Chowdhury, B.; Bhaumik, A. Catalytic reduction of CO2 into fuels and fine chemicals. Green Chem. 2020, 22, 4002–4033.
- Etim, U.J.; Semiat, R.; Zhong, Z. CO2 Valorization Reactions over Cu-Based Catalysts: Characterization and the Nature of Active Sites. Am. J. Chem. Eng. 2021, 9, 53–78.
- Green, A.E.; Justen, J.; Schöllkopf, W.; Gentleman, A.S.; Fielicke, A.; Mackenzie, S.R. IR Signature of Size-Selective CO2 Activation on Small Platinum Cluster Anions, Ptn−(n= 4–7). Angew. Chem. 2018, 130, 15038–15042.
- Li, H.; Zhao, J.; Luo, L.; Du, J.; Zeng, J. Symmetry-Breaking Sites for Activating Linear Carbon Dioxide Molecules. Acc. Chem. Res. 2021, 54, 1454–1464.
- Mondal, K.; Banerjee, A.; Ghanty, T.K. Adsorption and activation of C on Zr n (n= 2–7) clusters. Phys. Chem. Chem. Phys. 2020, 22, 16877–16886.
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698.
- Dietz, L.; Piccinin, S.; Maestri, M. Mechanistic Insights into CO2 activation via reverse water–gas shift on metal surfaces. J. Phys. Chem. C 2015, 119, 4959–4966.
- Ko, J.; Kim, B.-K.; Han, J.W. Density functional theory study for catalytic activation and dissociation of CO2 on bimetallic alloy surfaces. J. Phys. Chem. C 2016, 120, 3438–3447.
- Wang, S.-G.; Liao, X.-Y.; Cao, D.-B.; Huo, C.-F.; Li, Y.-W.; Wang, J.; Jiao, H. Factors controlling the interaction of CO2 with transition metal surfaces. J. Phys. Chem. C 2007, 111, 16934–16940.
- Chang, X.; Wang, T.; Gong, J. CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci 2016, 9, 2177–2196.
- Ding, X.; De Rogatis, L.; Vesselli, E.; Baraldi, A.; Comelli, G.; Rosei, R.; Savio, L.; Vattuone, L.; Rocca, M.; Fornasiero, P. Interaction of carbon dioxide with Ni (110): A combined experimental and theoretical study. Phys. Rev. B 2007, 76, 195425.
- Eren, B.; Weatherup, R.S.; Liakakos, N.; Somorjai, G.A.; Salmeron, M. Dissociative carbon dioxide adsorption and morphological changes on Cu (100) and Cu (111) at ambient pressures. J. Am. Chem. Soc. 2016, 138, 8207–8211.
- Hammami, R.; Dhouib, A.; Fernandez, S.; Minot, C. CO2 adsorption on (0 0 1) surfaces of metal monoxides with rock-salt structure. Catal. Today 2008, 139, 227–233.
- Solymosi, F. The bonding, structure and reactions of CO2 adsorbed on clean and promoted metal surfaces. J. Mol. Catal. 1991, 65, 337–358.
- Mishra, A.K.; Roldan, A.; de Leeuw, N.H. CuO surfaces and CO2 activation: A dispersion-corrected DFT+ U study. J. Phys. Chem. C 2016, 120, 2198–2214.
- Yang, T.; Gu, T.; Han, Y.; Wang, W.; Yu, Y.; Zang, Y.; Zhang, H.; Mao, B.; Li, Y.; Yang, B. Surface orientation and pressure dependence of CO2 activation on Cu surfaces. J. Phys. Chem. C 2020, 124, 27511–27518.
- Yu, H.; Cao, D.; Fisher, A.; Johnston, R.L.; Cheng, D. Size effect on the adsorption and dissociation of CO2 on Co nanoclusters. Appl. Surf. Sci. 2017, 396, 539–546.
- Etim, U.J.; Song, Y.; Zhong, Z. Improving the Cu/ZnO-Based Catalysts for Carbon Dioxide Hydrogenation to Methanol, and the Use of Methanol As a Renewable Energy Storage Media. Front. Energy Res. 2020, 8, 545431.
- Fu, S.S.; Somorjai, G.A. Interactions of O2, CO, CO2, and D2 with the stepped Cu (311) crystal face: Comparison to Cu (110). Surf. Sci. 1992, 262, 68–76.
- Rasmussen, P.; Taylor, P.; Chorkendorff, I. The interaction of carbon dioxide with Cu (100). Surf. Sci. 1992, 269, 352–359.
- Muttaqien, F.; Hamamoto, Y.; Inagaki, K.; Morikawa, Y. Dissociative adsorption of CO2 on flat, stepped, and kinked Cu surfaces. J. Chem. Phys. 2014, 141, 034702.
- Pohl, M.; Otto, A. Adsorption and reaction of carbon dioxide on pure and alkali-metal promoted cold-deposited copper films. Surf. Sci. 1998, 406, 125–137.
- Wang, S.-G.; Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. Chemisorption of CO2 on nickel surfaces. J. Phys. Chem. B 2005, 109, 18956–18963.
- Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. CO2 dissociation on Ni (2 1 1). Surf. Sci. 2009, 603, 2991–2998.
- Illing, G.; Heskett, D.; Plummer, E.; Freund, H.-J.; Somers, J.; Lindner, T.; Bradshaw, A.; Buskotte, U.; Neumann, M.; Starke, U. Adsorption and reaction of CO2 on Ni : X-ray photoemission, near-edge X-ray absorption fine-structure and diffuse leed studies. Surf. Sci. 1988, 206, 1–19.
- Heine, C.; Lechner, B.A.; Bluhm, H.; Salmeron, M. Recycling of CO2: Probing the chemical state of the Ni (111) surface during the methanation reaction with ambient-pressure X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 2016, 138, 13246–13252.
- Roiaz, M.; Monachino, E.; Dri, C.; Greiner, M.; Knop-Gericke, A.; Schlögl, R.; Comelli, G.; Vesselli, E. Reverse water–gas shift or Sabatier methanation on Ni (110)? Stable surface species at near-ambient pressure. J. Am. Chem. Soc. 2016, 138, 4146–4154.
- Cai, J.; Han, Y.; Chen, S.; Crumlin, E.J.; Yang, B.; Li, Y.; Liu, Z. CO2 activation on Ni (111) and Ni (100) surfaces in the presence of H2O: An ambient-pressure X-ray photoelectron spectroscopy study. J. Phys. Chem. C 2019, 123, 12176–12182.
- Silaghi, M.-C.; Comas-Vives, A.; Coperet, C. CO2 Activation on Ni/γ–Al2O3 catalysts by first-principles calculations: From ideal surfaces to supported nanoparticles. ACS Catal. 2016, 6, 4501–4505.
- Liu, Z.; Zhou, Y.; Solymosi, F.; White, J. Spectroscopic study of K-induced activation of CO2 on Pt (111). Surf. Sci. 1991, 245, 289–304.
- Ricart, J.M.; Habas, M.P.; Clotet, A.; Curulla, D.; Illas, F. Theoretical study of CO2 activation on Pt (111) induced by coadsorbed K atoms. Surf. Sci. 2000, 460, 170–181.
- Huang, M.; Adnot, A.; Suppiah, S.; Kaliaguine, S. XPS observation of surface interaction between H2 and CO2 on platinum foil. J. Mol. Catal. A: Chem. 1995, 104, L131–L137.
- Su, H.; Ye, Y.; Lee, K.-J.; Zeng, J.; Mun, B.S.; Crumlin, E.J. Probing the surface chemistry for reverse water gas shift reaction on Pt (1 1 1) using ambient pressure X-ray photoelectron spectroscopy. J. Catal. 2020, 391, 123–131.
- Megha; Mondal, K.; Ghanty, T.K.; Banerjee, A. Adsorption and Activation of CO2 on Small-Sized Cu–Zr Bimetallic Clusters. J. Phys. Chem. A 2021, 125, 2558–2572.
- Niu, J.; Ran, J.; Ou, Z.; Du, X.; Wang, R.; Qi, W.; Zhang, P. CO2 dissociation over PtxNi4− x bimetallic clusters with and without hydrogen sources: A density functional theory study. J. CO2 Util. 2016, 16, 431–441.
- Jia, J.; Qian, C.; Dong, Y.; Li, Y.F.; Wang, H.; Ghoussoub, M.; Butler, K.T.; Walsh, A.; Ozin, G.A. Heterogeneous catalytic hydrogenation of CO2 by metal oxides: Defect engineering–perfecting imperfection. Chem. Soc. Rev. 2017, 46, 4631–4644.
- Seiferth, O.; Wolter, K.; Dillmann, B.; Klivenyi, G.; Freund, H.-J.; Scarano, D.; Zecchina, A. IR investigations of CO2 adsorption on chromia surfaces: Cr2O3 (0001)/Cr (110) versus polycrystalline α-Cr2O3. Surf. Sci. 1999, 421, 176–190.
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024.
- Liu, L.; Fan, W.; Zhao, X.; Sun, H.; Li, P.; Sun, L. Surface dependence of CO2 adsorption on Zn2GeO4. Langmuir 2012, 28, 10415–10424.
- Rodriguez, J.A.; Liu, P.; Stacchiola, D.J.; Senanayake, S.D.; White, M.G.; Chen, J.G. Hydrogenation of CO2 to methanol: Importance of metal–oxide and metal–carbide interfaces in the activation of CO2. ACS Catal. 2015, 5, 6696–6706.
- Hezam, A.; Namratha, K.; Drmosh, Q.A.; Ponnamma, D.; Wang, J.; Prasad, S.; Ahamed, M.; Cheng, C.; Byrappa, K. CeO2 nanostructures enriched with oxygen vacancies for photocatalytic CO2 reduction. ACS Appl. Nano Mater. 2019, 3, 138–148.
- Bu, Y.; Weststrate, C.; Niemantsverdriet, J.; Fredriksson, H.O. Role of ZnO and CeO x in Cu-Based Model Catalysts in Activation of H2O and CO2 Dynamics Studied by in Situ Ultraviolet–Visible and X-ray Photoelectron Spectroscopy. ACS Catal. 2016, 6, 7994–8003.
- Chen, S.; Cao, T.; Gao, Y.; Li, D.; Xiong, F.; Huang, W. Probing surface structures of CeO2, TiO2, and Cu2O nanocrystals with CO and CO2 chemisorption. J. Phys. Chem. C 2016, 120, 21472–21485.
- Wang, Y.; Zhao, J.; Wang, T.; Li, Y.; Li, X.; Yin, J.; Wang, C. CO2 photoreduction with H2O vapor on highly dispersed CeO2/TiO2 catalysts: Surface species and their reactivity. J. Catal. 2016, 337, 293–302.
- Di, J.; Zhu, C.; Ji, M.; Duan, M.; Long, R.; Yan, C.; Gu, K.; Xiong, J.; She, Y.; Xia, J. Defect-rich Bi12O17Cl2 nanotubes self-accelerating charge separation for boosting photocatalytic CO2 reduction. Angew. Chem. Int. Ed. 2018, 57, 14847–14851.
- Yan, S.C.; Ouyang, S.X.; Gao, J.; Yang, M.; Feng, J.Y.; Fan, X.X.; Wan, L.J.; Li, Z.S.; Ye, J.H.; Zhou, Y. A room-temperature reactive-template route to mesoporous ZnGa2O4 with improved photocatalytic activity in reduction of CO2. Angew. Chem. 2010, 122, 6544–6548.
- Guo, J.; Ouyang, S.; Kako, T.; Ye, J. Mesoporous In(OH)3 for photoreduction of CO2 into renewable hydrocarbon fuels. Appl. Surf. Sci. 2013, 280, 418–423.
- Cao, A.; Wang, Z.; Li, H.; Nørskov, J.K. Relations between Surface Oxygen Vacancies and Activity of Methanol Formation from CO2 Hydrogenation over In2O3 Surfaces. ACS Catal. 2021, 11, 1780–1786.
- Staudt, T.; Lykhach, Y.; Tsud, N.; Skála, T.S.; Prince, K.C.; Matolín, V.R.; Libuda, J.R. Electronic structure of magnesia–ceria model catalysts, CO2 adsorption, and CO2 activation: A synchrotron radiation photoelectron spectroscopy study. J. Phys. Chem. C 2011, 115, 8716–8724.
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Sanz, J.F. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–550.
- Cheng, Z.; Sherman, B.J.; Lo, C.S. Carbon dioxide activation and dissociation on ceria (110): A density functional theory study. J. Chem. Phys. 2013, 138, 014702.
- Yang, C.; Bebensee, F.; Chen, J.; Yu, X.; Nefedov, A.; Wöll, C. Carbon dioxide adsorption on CeO2 (110): An XPS and NEXAFS study. ChemPhysChem 2017, 18, 1874–1880.
- Hahn, K.R.; Iannuzzi, M.; Seitsonen, A.P.; Hutter, J.R. Coverage effect of the CO2 adsorption mechanisms on CeO2 (111) by first principles analysis. J. Phys. Chem. C 2013, 117, 1701–1711.
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638.
- Thompson, T.L.; Diwald, O.; Yates, J.T. CO2 as a probe for monitoring the surface defects on TiO2 (110) temperature-programmed desorption. J. Phys. Chem. B 2003, 107, 11700–11704.
- Funk, S.; Burghaus, U. Adsorption of CO2 on oxidized, defected, hydrogen and oxygen covered rutile (1 × 1)-TiO2 (110). Phys. Chem. Chem. Phys. 2006, 8, 4805–4813.
- Suriye, K.; Praserthdam, P.; Jongsomjit, B. Control of Ti3+ surface defect on TiO2 nanocrystal using various calcination atmospheres as the first step for surface defect creation and its application in photocatalysis. Appl. Surf. Sci. 2007, 253, 3849–3855.
- Suriye, K.; Jongsomjit, B.; Satayaprasert, C.; Praserthdam, P. Surface defect (Ti3+) controlling in the first step on the anatase TiO2 nanocrystal by using sol–gel technique. Appl. Surf. Sci. 2008, 255, 2759–2766.
- Sorescu, D.C.; Al-Saidi, W.A.; Jordan, K.D. CO2 adsorption on TiO2 (101) anatase: A dispersion-corrected density functional theory study. J. Chem. Phys. 2011, 135, 124701.
- Liao, L.-F.; Lien, C.-F.; Shieh, D.-L.; Chen, M.-T.; Lin, J.-L. FTIR study of adsorption and photoassisted oxygen isotopic exchange of carbon monoxide, carbon dioxide, carbonate, and formate on TiO2. J. Phys. Chem. B 2002, 106, 11240–11245.
- Lee, J.; Sorescu, D.C.; Deng, X. Electron-induced dissociation of CO2 on TiO2 (110). J. Am. Chem. Soc. 2011, 133, 10066–10069.
- Nolan, M.; Fronzi, M. Activation of CO2 at chromia-nanocluster-modified rutile and anatase TiO2. Catal. Today 2019, 326, 68–74.
- Nolan, M. Adsorption of CO2 on heterostructures of Bi2O3 nanocluster-modified TiO2 and the role of reduction in promoting CO2 activation. ACS Omega 2018, 3, 13117–13128.
- Pavelec, J.; Hulva, J.; Halwidl, D.; Bliem, R.; Gamba, O.; Jakub, Z.; Brunbauer, F.; Schmid, M.; Diebold, U.; Parkinson, G.S. A multi-technique study of CO2 adsorption on Fe3O4 magnetite. J. Chem. Phys. 2017, 146, 014701.
- Hakim, A.; Marliza, T.S.; Abu Tahari, N.M.; Wan Isahak, R.W.; Yusop, R.M.; Mohamed Hisham, W.M.; Yarmo, A.M. Studies on CO2 adsorption and desorption properties from various types of iron oxides (FeO, Fe2O3, and Fe3O4). Ind. Eng. Chem. Res. 2016, 55, 7888–7897.
- Li, X.; Paier, J. Vibrational properties of CO2 adsorbed on the Fe3O4 (111) surface: Insights gained from DFT. J. Chem. Phy 2020, 152, 104702.
- Mirabella, F.; Zaki, E.; Ivars-Barcelo, F.; Schauermann, S.; Shaikhutdinov, S.; Freund, H.-J. CO2 adsorption on magnetite Fe3O4 (111). J. Phys. Chem. C 2018, 122, 27433–27441.
- Su, T.; Qin, Z.; Huang, G.; Ji, H.; Jiang, Y.; Chen, J. Density functional theory study on the interaction of CO2 with Fe3O4 (111) surface. Appl. Surf. Sci. 2016, 378, 270–276.
- Tang, Q.-L.; Hong, Q.-J.; Liu, Z.-P. CO2 fixation into methanol at Cu/ZrO2 interface from first principles kinetic Monte Carlo. J. Catal. 2009, 263, 114–122.
- Fisher, I.A.; Bell, A.T. In-situinfrared study of methanol synthesis from H2/CO2 over Cu/SiO2 and Cu/ZrO2/SiO2. J. Catal. 1997, 172, 222–237.