Alzheimer’s disease (AD) is the most common neurodegenerative disease, and globally, over 46 million people are affected by this devastating disease. AD causes irreversible mental and cognitive deficiency including memory loss, personality disorder, and intellectual abnormality in patients older than 65 years. Central sensory systems including the visual system are also affected during the advanced stages of the disease . Collectively, complications of AD diminish the lifespan, hamper the quality of life, and cause physical impairment, which finally appears as a terrible difficulty in normal life activities. To decrease the social and economic costs and the burden of the disease on patients and their families, recently, several attempts have been made to identify disease diagnostic markers. Neuroimaging methods, such as magnetic resonance imaging and positron emission tomography, have been developed enabling researchers to diagnose AD at the early stages of the disease. Moreover, distinct biomarkers, which are essential to figure out the pathological characteristics of AD, have been observed in the cerebrospinal fluid (CSF). The advancement of AD is associated with three cardinal neuropathological features such as extracellular deposition of amyloid-β (Aβ) to produce neuritic plaques, intracellular neurofibrillary tangles (NFTs) and synaptic degeneration. These pathological alterations arise in the neocortex, hippocampus, and other subcortical regions that are crucial for cognitive functions. Aβ has been considered as the foremost risk factor that playing a vital role in the initiation and progression of AD. Aβ is generated into the typical individual, but in some instances, this peptide leads to aggregation, the starting point of disease progression. Many findings elucidate that Aβ oligomers might play a central role in neuronal dysfunction and AD.
- Amyloid Precursor Protein
- Amyloid-β
- Alzheimer’s Disease
1. Introduction
The amyloid precursor protein (APP) is a type I transmembrane glycoprotein containing 695–770 amino acids [1]. It is regarded that abnormal proteolytic processing of APP leads to the generation of Aβ [2]. In the non-amyloidogenic pathways, APP is cleaved by α- and γ-secretases. It is known that α-secretase is a metalloprotease (ADAM) which is localized to the Golgi complex and the cell membrane. This α-secretase can cleave APP at residue L688, located in the middle of the Aβ domain [3]. α-secretase causes APP cleavage leading to the formation of soluble APP alpha (sAPPα) and a cell-membrane-bound C-terminal fragment 83 (CTF83). The generated CTF83 is cleaved by γ-secretase to produce AICD and a small p3 fragment.
In the amyloidogenic pathways, APP is cleaved by β- and γ-secretases [50]. β-secretase (BACE1) belongs to the aspartic protease family and is around 500 residues in length containing 2 active sites situated at the lumenal side of the membrane [52]. The β-secretase enzyme possesses high sequence specificity [53,54], a feature that matches with BACE1, which can cleave APP at theIn the amyloidogenic pathways, APP is cleaved by β- and γ-secretases [1]. β-secretase (BACE1) belongs to the aspartic protease family and is around 500 residues in length containing 2 active sites situated at the lumenal side of the membrane [3]. The β-secretase enzyme possesses high sequence specificity [4][5], a feature that matches with BACE1, which can cleave APP at the
N-terminal of the Aβ domain, either at residues Glu682 or Asp672 [52]. BACE1 also causes APP cleavage and generates soluble APP β (sAPPβ) and a cell-membrane-bound C-terminal fragment 99 (CTF99).-terminal of the Aβ domain, either at residues Glu682 or Asp672 [3]. BACE1 also causes APP cleavage and generates soluble APP β (sAPPβ) and a cell-membrane-bound C-terminal fragment 99 (CTF99).
On the other hand, γ-secretase is an enzymatic complex of four proteins including presenilin enhancer 2, presenilin (PSEN), anterior pharynx defective 1, and nicastrin. Indeed, each of the subunits is considered as an effective therapeutic for elevating Aβ clearance or controlling Aβ generation. Within the membrane, γ-secretase causes cleavage of βCTF99 that leads to the formation of the APP intracellular domain (AICD) and Aβ peptides. Various isoforms of Aβ isoforms are available that contain a variable length of amino acid residues including AβOn the other hand, γ-secretase is an enzymatic complex of four proteins including presenilin enhancer 2, presenilin (PSEN), anterior pharynx defective 1, and nicastrin. Indeed, each of the subunits is considered as an effective therapeutic for elevating Aβ clearance or controlling Aβ generation. Within the membrane, γ-secretase causes cleavage of βCTF99 that leads to the formation of the APP intracellular domain (AICD) and Aβ peptides. Various isoforms of Aβ isoforms are available that contain a variable length of amino acid residues including Aβ
1–40(i.e., the most abundant) and Aβ
1–42(i.e., the less soluble isoform). Aggregation of Aβ triggers the generation of protofibrils, fibrils, and oligomers, which eventually can lead to the formation of plaques, which are well-known as one of the major hallmarks of AD pathology as shown in
Figure 1.
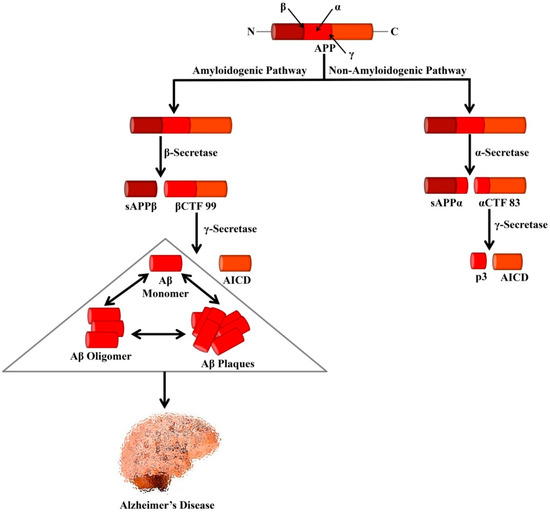
Processing of APP by secretases that leads to the formation of Alzheimer’s-associated Aβ peptides. In the amyloidogenic pathway, APP is cleaved by β- and γ-secretases leading to the formation of Aβ peptides and AICD. Formation of the neurotoxic Aβ is the major cause of AD. In the non-amyloidogenic pathway, APP is cleaved by α- and γ-secretases leading to the genesis of p3 and AICD. APP, Amyloid precursor protein; sAPPα, Soluble APP alpha; sAPPβ, Soluble APP beta; αCTF 83, Alpha C-terminal fragment 83; βCTF 99, Beta C-terminal fragment 99; AICD, APP intracellular domain.
It is believed that in the AD process, Aβ accumulation in the brain is one of the events that take place initially [6]. Accumulation of Aβ also initiates in the entorhinal cortex and hippocampus. Furthermore, in NFTs, intracellular hyperphosphorylated tau deposition can cause disruption in axonal transport and progressive alterations in the cytoskeleton. In 1991, several independent groups initially proposed the theory that Aβ accumulation is the major characteristic of AD pathogenesis [7][8][9]. After one year, Hardy and Higgins [10] formally proposed the ‘amyloid cascade hypothesis’. As per the hypothesis, the deposition of Aβ in the brain can trigger several events including cognitive deficit, neuronal death, synaptic loss, formation of NFTs, and phosphorylation of tau.
It is believed that in the AD process, Aβ accumulation in the brain is one of the events that take place initially [55]. Accumulation of Aβ also initiates in the entorhinal cortex and hippocampus. Furthermore, in NFTs, intracellular hyperphosphorylated tau deposition can cause disruption in axonal transport and progressive alterations in the cytoskeleton. In 1991, several independent groups initially proposed the theory that Aβ accumulation is the major characteristic of AD pathogenesis [56,57,58]. After one year, Hardy and Higgins [59] formally proposed the ‘amyloid cascade hypothesis’. As per the hypothesis, the deposition of Aβ in the brain can trigger several events including cognitive deficit, neuronal death, synaptic loss, formation of NFTs, and phosphorylation of tau. The amyloid cascade hypothesis was further strengthened by the fact that AD may take place due to the autosomal dominant mutations in theThe amyloid cascade hypothesis was further strengthened by the fact that AD may take place due to the autosomal dominant mutations in the
APPgene and these mutations in
PSEN1and
PSEN2 can elevate the Aβ generation and ultimately mediate the generation of Aβ aggregates and deposits [60]. Transgenic mouse models that express forms of PSEN proteins or APP containing mutations linked with human FAD progressively show the development of memory impairments and Aβ plaques in the brain, which further strengthens the hypothesis that buildup of Aβ can trigger AD [61]. Mutations incan elevate the Aβ generation and ultimately mediate the generation of Aβ aggregates and deposits [11]. Transgenic mouse models that express forms of PSEN proteins or APP containing mutations linked with human FAD progressively show the development of memory impairments and Aβ plaques in the brain, which further strengthens the hypothesis that buildup of Aβ can trigger AD [12]. Mutations in
PSENseem to be the major cause of FAD with over 150 causative mutations that have been mapped to the genes (
PSEN1and
PSEN2) encoding the PSEN proteins [62,63]. Most of these mutations seem to elevate the generation of Aβ) encoding the PSEN proteins [13][14]. Most of these mutations seem to elevate the generation of Aβ
1–42over Aβ
1–40 by mediating cleavage at residue 639 of APP over residue 637 [64]. Increased activity of BACE1 [65] and defective clearance [66] are found to contribute to the buildup of Aβ in the brain in late-onset sporadic AD (SAD). The buildup of Aβ is also found to be associated with the apolipoprotein E ε4 (by mediating cleavage at residue 639 of APP over residue 637 [15]. Increased activity of BACE1 [16] and defective clearance [17] are found to contribute to the buildup of Aβ in the brain in late-onset sporadic AD (SAD). The buildup of Aβ is also found to be associated with the apolipoprotein E ε4 (
APOE ε4) allele, which is regarded as the strongest genetic risk factor for late-onset SAD.
It has been revealed by post-mortem analyses that healthy elderly individuals can possess extensive amyloid pathology [18]. Moreover, brain imaging analyses showed that Aβ pathology was observed in up to 44% of cognitively healthy older people [19]. As compared to the individuals without amyloid pathology, a faster deficit in brain glucose metabolism, brain volume, and cognitive performance was experienced by individuals with amyloid deposits at baseline [20][21][22][23][24]. By 15–20 years, it has been estimated that the deposition of Aβ will precede the clinical AD symptoms [25].
It has been revealed by post-mortem analyses that healthy elderly individuals can possess extensive amyloid pathology [67]. Moreover, brain imaging analyses showed that Aβ pathology was observed in up to 44% of cognitively healthy older people [68]. As compared to the individuals without amyloid pathology, a faster deficit in brain glucose metabolism, brain volume, and cognitive performance was experienced by individuals with amyloid deposits at baseline [69,70,71,72,73]. By 15–20 years, it has been estimated that the deposition of Aβ will precede the clinical AD symptoms [70].References
- Uddin, M.S.; Kabir, M.T.; Jeandet, P.; Mathew, B.; Ashraf, G.M.; Perveen, A.; Bin-Jumah, M.N.; Mousa, S.A.; Abdel-Daim, M.M. Novel Anti-Alzheimer’s Therapeutic Molecules Targeting Amyloid Precursor Protein Processing. Oxid. Med. Cell. Longev. 2020, 2020, 1–19.
- Weggen, S.; Beher, D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimer’s Res. Ther. 2012, 4, 1–14.
- MacLeod, R.; Hillert, E.-K.; Cameron, R.T.; Baillie, G.S. The role and therapeutic targeting of α-, β- and γ-secretase in Alzheimer’s disease. Futur. Sci. OA 2015, 1, FSO11.
- Citron, M.; Teplow, D.B.; Selkoe, D.J. Generation of amyloid β protein from its precursor is sequence specific. Neuron 1995, 14, 661–670.
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the β-amyloid precursor protein in familial Alzheimer’s disease increases β-protein production. Nature 1992, 360, 672–674.
- Uddin, M.S.; Kabir, M.T.; Rahman, M.M.; Mathew, B.; Shah, M.A.; Ashraf, G.M. TV 3326 for Alzheimer’s dementia: A novel multimodal ChE and MAO inhibitors to mitigate Alzheimer’s-like neuropathology. J. Pharm. Pharmacol. 2020, 72, 1001–1012.
- Beyreuther, K.; Masters, C.L. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: Precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1991, 1, 241–251.
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388.
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498.
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185.
- Kabir, M.T.; Uddin, M.S.; Setu, J.R.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Exploring the Role of PSEN mutations in the pathogenesis of Alzheimer’s disease. Neurotox. Res. 2020, doi:10.1007/s12640-020-00232-x.
- Karran, E.; Mercken, M.; Strooper, B. De The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712.
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin I variants elevate aβ1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013.
- Ertekin-Taner, N. Genetics of Alzheimer’s Disease: A Centennial Review. Neurol. Clin. 2007, 25, 611–667.
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946.
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease [1]. Nat. Med. 2003, 9, 3–4.
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774.
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Kelly, J.F.; Aggarwal, N.T.; Shah, R.C.; Wilson, R.S. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006, 66, 1837–1844.
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.J.; Visser, P.J.; Aalten, P.; Aarsland, D.; Alcolea, D.; et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA—J. Am. Med. Assoc. 2015, 313, 1924–1938.
- Vos, S.J.B.; Xiong, C.; Visser, P.J.; Jasielec, M.S.; Hassenstab, J.; Grant, E.A.; Cairns, N.J.; Morris, J.C.; Holtzman, D.M.; Fagan, A.M. Preclinical Alzheimer’s disease and its outcome: A longitudinal cohort study. Lancet Neurol. 2013, 12, 957–965.
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367.
- Burnham, S.C.; Bourgeat, P.; Doré, V.; Savage, G.; Brown, B.; Laws, S.; Maruff, P.; Salvado, O.; Ames, D.; Martins, R.N.; et al. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer’s disease pathophysiology (SNAP) or Alzheimer’s disease pathology: A longitudinal study. Lancet Neurol. 2016, 15, 1044–1053.
- Petersen, R.C.; Wiste, H.J.; Weigand, S.D.; Rocca, W.A.; Roberts, R.O.; Mielke, M.M.; Lowe, V.J.; Knopman, D.S.; Pankratz, V.S.; Machulda, M.M.; et al. Association of elevated amyloid levels with cognition and biomarkers in cognitively normal people from the community. JAMA Neurol. 2016, 73, 85–92.
- Donohue, M.C.; Sperling, R.A.; Petersen, R.; Sun, C.K.; Weiner, M.; Aisen, P.S. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA—J. Am. Med. Assoc. 2017, 317, 2305–2316.
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255.