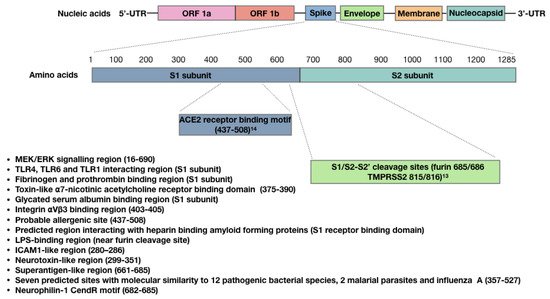
The main host target receptor for the SARS-CoV-2 spike protein is angiotensin-converting enzyme 2 (ACE2), which is involved in maintaining blood pressure and vascular remodeling, and is expressed on adipocytes, other cells at mucosal surfaces, and in the vasculature, heart, kidneys, pancreas and brain.
1. Bioactivity of the SARS-CoV-2 Spike Protein
ACE2 is also expressed within placental tissues [1], and is involved in regulating fetal myocardial growth and lung and brain development [2]. A recent pre-print study showed that blocking ACE2 with an anti-ACE2 antibody reduced placental SARS-CoV-2 infection [3]. This is one of a number of studies that have demonstrated that the placenta is susceptible to SARS-CoV-2 [2][4], and may also be responsive to spike proteins, which have been identified at low concentrations in plasma from recipients of the Moderna vaccine [5].
A large number of studies have provided evidence that the SARS-CoV-2 spike protein is bioactive, and that ligation of the spike protein to ACE2 explains some of its bioactivity. A study by Lei et al. [6], for example, demonstrated that the spike protein down-regulated ACE2 in Syrian hamster vascular endothelial cells, which led to inhibited mitochondrial function and cell damage. In a later in vitro study, however, the spike protein was shown to upregulate bronchial epithelial cell ACE2 expression via activation of the Type I IFN signaling pathway [7]. These two studies indicate that the effect of this spike protein on ACE2 expression is tissue or species-specific. A recent study using mice and human umbilical cord blood demonstrated that ligation of recombinant spike protein to ACE2 can activate Nlrp3 inflammasome assembly, resulting in uncontrolled inflammation leading to pyroptotic cell death [8]. Ropa et al. [9] demonstrated that hematopoietic stem cells from human umbilical cord blood express ACE2 and were adversely affected by spike protein in terms of their ability to proliferate and expand into progenitor cells. Ropa et al. proposed that this could explain the reduced numbers of circulating lymphocytes and platelets that are observed in COVID-19 patients [9]. Using wild type and transgenic mice expressing human ACE2, Colunga et al. [10] recently demonstrated that intratracheal administration of the spike protein S1 subunit induced alveolar inflammation and acute lung injury and altered lung vascular permeability, leading to an ACE2-dependent systemic cytokine storm. Suzuki et al. recently demonstrated that the spike protein S1 subunit (Val-16-Gln-690), but not the ACE2 receptor-binding domain (Arg-319-Phe-541), elicited mitogen-activated protein kinase (MEK/ERK) signaling in human pulmonary artery smooth muscle and endothelial cells [11]. These authors proposed that this growth factor/hormone-like cell signaling contributes to the hyperplasia and/or hypertrophy of vascular smooth muscle and endothelial cells in patients with COVID-19 and may also possibly explain some of the AVRs associated with COVID-19 vaccines [11]. By combining their knowledge of the SARS-CoV-2 spike protein, and the work of Chen et al. [12] on the SARS-CoV-1 spike protein conducted on human pneumocytes, Suzuki proposed that the spike protein functionally converts ACE2 from a peptidase to a functional cell membrane signaling receptor (
ACE2 is also expressed within placental tissues [52], and is involved in regulating fetal myocardial growth and lung and brain development [53]. A recent pre-print study showed that blocking ACE2 with an anti-ACE2 antibody reduced placental SARS-CoV-2 infection [54]. This is one of a number of studies that have demonstrated that the placenta is susceptible to SARS-CoV-2 [53,55], and may also be responsive to spike proteins, which have been identified at low concentrations in plasma from recipients of the Moderna vaccine [56].
A large number of studies have provided evidence that the SARS-CoV-2 spike protein is bioactive, and that ligation of the spike protein to ACE2 explains some of its bioactivity. A study by Lei et al. [57], for example, demonstrated that the spike protein down-regulated ACE2 in Syrian hamster vascular endothelial cells, which led to inhibited mitochondrial function and cell damage. In a later in vitro study, however, the spike protein was shown to upregulate bronchial epithelial cell ACE2 expression via activation of the Type I IFN signaling pathway [58]. These two studies indicate that the effect of this spike protein on ACE2 expression is tissue or species-specific. A recent study using mice and human umbilical cord blood demonstrated that ligation of recombinant spike protein to ACE2 can activate Nlrp3 inflammasome assembly, resulting in uncontrolled inflammation leading to pyroptotic cell death [59]. Ropa et al. [60] demonstrated that hematopoietic stem cells from human umbilical cord blood express ACE2 and were adversely affected by spike protein in terms of their ability to proliferate and expand into progenitor cells. Ropa et al. proposed that this could explain the reduced numbers of circulating lymphocytes and platelets that are observed in COVID-19 patients [60]. Using wild type and transgenic mice expressing human ACE2, Colunga et al. [61] recently demonstrated that intratracheal administration of the spike protein S1 subunit induced alveolar inflammation and acute lung injury and altered lung vascular permeability, leading to an ACE2-dependent systemic cytokine storm. Suzuki et al. recently demonstrated that the spike protein S1 subunit (Val-16-Gln-690), but not the ACE2 receptor-binding domain (Arg-319-Phe-541), elicited mitogen-activated protein kinase (MEK/ERK) signaling in human pulmonary artery smooth muscle and endothelial cells [62]. These authors proposed that this growth factor/hormone-like cell signaling contributes to the hyperplasia and/or hypertrophy of vascular smooth muscle and endothelial cells in patients with COVID-19 and may also possibly explain some of the AVRs associated with COVID-19 vaccines [62]. By combining their knowledge of the SARS-CoV-2 spike protein, and the work of Chen et al. [63] on the SARS-CoV-1 spike protein conducted on human pneumocytes, Suzuki proposed that the spike protein functionally converts ACE2 from a peptidase to a functional cell membrane signaling receptor ( Figure 1).
Figure 1. Sites within the SARS-CoV-2 spike protein S1 subunit and receptor binding domain, showing confirmed or predicted bioactivity.
Sites within the SARS-CoV-2 spike protein S1 subunit and receptor binding domain, showing confirmed or predicted bioactivity.
A number of studies have shown that the SARS-CoV-2 spike protein also possesses ACE2-independent bioactivity. Nader et al. [64] for example, found that in addition to ACE2, SARS-CoV-2 can also attach to, invade and damage host cells via αVβ3 integrin adhesion molecules, which are highly expressed on vascular endothelial cells. These authors demonstrated that an arginine–glycine–aspartic acid mutation (RGD motif) in this spike protein has uniquely allowed SARS-CoV-2 to acquire this function. Since the RGD motif is located adjacent to the ACE2 receptor-binding motif (Figure 1), this could allow SARS-CoV-2 to bind to cells lacking ACE2 and to potentially enhance binding to cells expressing both ACE2 and αVβ3 integrin. Interestingly, αVβ3 integrins are also expressed on platelets and contribute to platelet activation and aggregation [65]. Shen et al. showed that SARS-CoV-2 interacts with platelets to influence their function and promote dysregulated coagulation [66]; they proposed an ACE2-independent mechanism for this, because the expression of ACE2 is uncertain in platelets and their progenitor megakaryocytes [67]. It is possible that αVβ3 integrin is involved in SARS-CoV-2 interactions with platelets.
In silico analysis of the S1 subunit of the SARS-CoV-2 spike protein has revealed molecular docking sites for TLRs, including TLR1, TLR4 and TLR6; interactions with spike protein were the strongest for TLR4 [68] (Figure 1). An in vitro study performed by Shirato and Kizaki [69] demonstrated that the spike protein S1 subunit induced murine peritoneal macrophages to secrete pro-inflammatory cytokines via TLR4 signaling and that the response was attenuated using a TLR4 antagonist. TLR4 is also highly expressed in platelets, and when bacterial lipopolysaccharide (LPS) binds to TLR4, it can result in thrombocytopenia and the accumulation of platelets in the lungs [70]. Ouyang et al. [71] recently demonstrated that SARS-CoV-2 spike protein can also bind to bacterial LPS, and this spike protein-LPS interaction was shown to boost monocyte NF-κB activation and cytokine responses in vitro, as well as NF-κB activation in vivo [72]. Petruk et al. predicted the LPS interacting region to be within the proximity of the spike protein S1/S2 furin cleavage site (Figure 1), and proposed that spike protein–LPS interactions may in part explain the increased risk of severe COVID-19 caused by comorbidities.
The SARS-CoV-2 spike protein can also interact with other proteins. Grobbelaar et al. [73] demonstrated that when the spike protein S1 subunit was added to platelet-poor plasma, it interacted with and structurally modified plasma proteins β and γ fibrinogen, complement 3 and prothrombin, which made them more resistant to trypsinization. These authors proposed that this may contribute to the hypercoagulation associated with COVID-19 and may impair clot breakdown during fibrinolysis (Figure 1). The SARS-CoV-2 spike protein can also bind with high affinity to glycated human serum albumin. This may allow SARS-CoV-2 to evade the detection of its receptor-binding domain (RBD) by neutralizing antibodies (Figure 1); however, it can also lead to albumin depletion and may contribute to fluid tissue–vascular imbalance that can give rise to septic shock [74]. Also found within the RBD of SARS-CoV-2 and SARS-CoV-1 spike proteins is a “toxin-like” epitope that shares homology to snake venom α-bungarotoxin [75], which is a highly specific blocker of nicotinic acetylcholine receptors. Lagoumintzis et al. [75] hypothesize that the SARS-CoV-2 spike protein may block the cholinergic anti-inflammatory pathway, allowing for uncontrolled inflammation to occur during COVID-19.
The SARS-CoV-2 spike protein can also bind to the b1b2 domain of the neuropilin-1 receptor (NRP-1) [76], which normally interacts with vascular endothelial growth factor-A (VEGF-A) in neurons. ACE2 is not present in most neurons [77], although reports of neurological symptoms are common in COVID-19 patients [78]. Interestingly, interactions between the polybasic 682RRAR685 amino acid sequence, termed the “C-end rule” (CendR) motif (Figure 1), with NRP-1 potentiates SARS-CoV-2 entry into host cells [79]. This CendR motif is not conserved in either SARS-CoV-1 or Middle East respiratory syndrome coronavirus (MERS-CoV), and it is hypothesized that a “silencing” of pain through subversion of VEGF-A/NRP-1 signaling may underlie increased disease transmission in asymptomatic individuals [80].
There are three other regions of interest within the spike protein RBD that may also contribute to spike protein bioactivity. The first region is predicted with high probability to be an allergenic sequence [29]. This region could therefore contribute to anaphylaxis in some patients that have received viral vector and/or mRNA COVID-19 vaccines. The second RBD region of interest potentially allows the spike protein to bind to amyloid-forming heparin-binding proteins, which could lead to accelerated aggregation of amyloid proteins within the brain [81]. This supports Classen’s concern that COVID-19 vaccines could potentially induce prion disease [82]. The third region of interest within the RBD contains seven predicted molecular sites that share similarities to different toxins or virulence factors from 12 different bacterial species, 2 malarial parasites and influenza A [83] (Figure 1).
There is one final aspect of this spike protein that warrants consideration regarding its bioactivity, and this stems from the hypothesis that COVID-19-associated multi-system inflammatory syndrome in children (MIS-C) and the cytokine storm observed in adult patients with severe COVID-19 is mediated by spike protein superantigenic activity. Rivas et al. [84] have built on this hypothesis, first by drawing parallels between these two COVID-19 conditions and toxic shock syndrome (TSS). The superantigen Staphylococcus Enterotoxin B (SEB), associated with TSS, is a biotoxin that causes polyclonal T-cell activation and proliferation, which leads to massive production of pro-inflammatory cytokines. These researchers used structure-based computer modelling to discover an SEB-like sequence (glutamic acid661–arginine685) near the spike protein S1/S2 cleavage site that exhibits high binding affinity to both the T-cell receptor (TCR) β chain and co-stimulatory molecule CD28 [85] (Figure 1). They also identified several neurotoxin-like sequences within the spike protein; one (threonine299–tyrosine351) also displayed a high tendency to bind to the TCR, and another is an ICAM1-like region (aparagine280–threonine286) that is predicted to stabilize interactions between the spike protein and the TCR. These researchers also demonstrated TCRV/β skewing of the T cell response in COVID-19 patients with more severe and hyper-inflammatory clinical courses, which is consistent with spike protein superantigen activity. Additionally, they showed that the SARS-CoV-2 mutation aspartic acid839–tyrosine predictably enhanced binding affinity of the spike protein to the TCR, and later this group also provided evidence that a repurposed anti-SEB antibody could prevent SARS-CoV-2 infection in vitro [86].
Collectively, the diverse bioactivity of the SARS-CoV-2 spike protein makes this an ideal target for the immune system to neutralize the virus, and all the current COVID-19 vaccine platforms have focused on this spike protein because it is highly immunogenic [87]. However, we should also be cognizant of these bioactive properties when designing COVID-19 vaccines to ensure that only nontoxic immunogenic portions of the spike protein are expressed, and that their expression is both temporally and spatially limited and does not provide selection pressure driving viral mutation. The mRNA vaccines have been designed to allow a host cell to express the spike protein in its cell membrane [88], and the expression of the spike protein throughout the body is dependent on the biodistribution of LNPs—which primarily relocate to the spleen and liver, but have also been found in various other tissues [16,19]. We currently have no idea how long spike proteins are expressed by different host cells and in what tissues spike protein expression can occur because biodistribution studies on the spike protein have not been carried out to date [19]. The mRNA sequence has also been modified by manufacturers, with the addition of proline residues at positions 986 and 987, which could allow them to reside longer in the plasma membrane [16]. A recent pre-print by Patterson et al. [89] indicated that a subset of monocytes from COVID-19 patients contained SARS-CoV-2 S1 mRNA and proteins for as long as 15 months post-acute infection. This raises the possibility of the spike protein being expressed by maternal immune cells in colostrum and milk from COVID-19 positive mothers; thus, the biodistribution of spike mRNA and protein could be especially relevant during lactation. A recent study by Golan et al. [90] suggested that biodistribution of mRNA to milk during lactation is not a concern, as none was detected in milk from 6 mothers 4–48 h post-Pfizer–BioNTech and Moderna vaccination. However, a study demonstrated that following COVID-19 mRNA vaccination, exosomes expressing spike protein could be detected in plasma up to 4 months post-vaccination [91], which is concerning because we, and others [92,93], have shown that exosomes can be shed in bodily fluids such as colostrum and milk.
A number of studies have shown that the SARS-CoV-2 spike protein also possesses ACE2-independent bioactivity. Nader et al. [13] for example, found that in addition to ACE2, SARS-CoV-2 can also attach to, invade and damage host cells via αVβ3 integrin adhesion molecules, which are highly expressed on vascular endothelial cells. These authors demonstrated that an arginine–glycine–aspartic acid mutation (RGD motif) in this spike protein has uniquely allowed SARS-CoV-2 to acquire this function. Since the RGD motif is located adjacent to the ACE2 receptor-binding motif (Figure 1), this could allow SARS-CoV-2 to bind to cells lacking ACE2 and to potentially enhance binding to cells expressing both ACE2 and αVβ3 integrin. Interestingly, αVβ3 integrins are also expressed on platelets and contribute to platelet activation and aggregation [14]. Shen et al. showed that SARS-CoV-2 interacts with platelets to influence their function and promote dysregulated coagulation [15]; they proposed an ACE2-independent mechanism for this, because the expression of ACE2 is uncertain in platelets and their progenitor megakaryocytes [16]. It is possible that αVβ3 integrin is involved in SARS-CoV-2 interactions with platelets.
In silico analysis of the S1 subunit of the SARS-CoV-2 spike protein has revealed molecular docking sites for TLRs, including TLR1, TLR4 and TLR6; interactions with spike protein were the strongest for TLR4 [17] (Figure 1). An in vitro study performed by Shirato and Kizaki [18] demonstrated that the spike protein S1 subunit induced murine peritoneal macrophages to secrete pro-inflammatory cytokines via TLR4 signaling and that the response was attenuated using a TLR4 antagonist. TLR4 is also highly expressed in platelets, and when bacterial lipopolysaccharide (LPS) binds to TLR4, it can result in thrombocytopenia and the accumulation of platelets in the lungs [19]. Ouyang et al. [20] recently demonstrated that SARS-CoV-2 spike protein can also bind to bacterial LPS, and this spike protein-LPS interaction was shown to boost monocyte NF-κB activation and cytokine responses in vitro, as well as NF-κB activation in vivo [21]. Petruk et al. predicted the LPS interacting region to be within the proximity of the spike protein S1/S2 furin cleavage site (Figure 1), and proposed that spike protein–LPS interactions may in part explain the increased risk of severe COVID-19 caused by comorbidities.
The SARS-CoV-2 spike protein can also interact with other proteins. Grobbelaar et al. [22] demonstrated that when the spike protein S1 subunit was added to platelet-poor plasma, it interacted with and structurally modified plasma proteins β and γ fibrinogen, complement 3 and prothrombin, which made them more resistant to trypsinization. These authors proposed that this may contribute to the hypercoagulation associated with COVID-19 and may impair clot breakdown during fibrinolysis (Figure 1). The SARS-CoV-2 spike protein can also bind with high affinity to glycated human serum albumin. This may allow SARS-CoV-2 to evade the detection of its receptor-binding domain (RBD) by neutralizing antibodies (Figure 1); however, it can also lead to albumin depletion and may contribute to fluid tissue–vascular imbalance that can give rise to septic shock [23]. Also found within the RBD of SARS-CoV-2 and SARS-CoV-1 spike proteins is a “toxin-like” epitope that shares homology to snake venom α-bungarotoxin [24], which is a highly specific blocker of nicotinic acetylcholine receptors. Lagoumintzis et al. [24] hypothesize that the SARS-CoV-2 spike protein may block the cholinergic anti-inflammatory pathway, allowing for uncontrolled inflammation to occur during COVID-19.
The SARS-CoV-2 spike protein can also bind to the b1b2 domain of the neuropilin-1 receptor (NRP-1) [25], which normally interacts with vascular endothelial growth factor-A (VEGF-A) in neurons. ACE2 is not present in most neurons [26], although reports of neurological symptoms are common in COVID-19 patients [27]. Interestingly, interactions between the polybasic 682RRAR685 amino acid sequence, termed the “C-end rule” (CendR) motif (Figure 1), with NRP-1 potentiates SARS-CoV-2 entry into host cells [28]. This CendR motif is not conserved in either SARS-CoV-1 or Middle East respiratory syndrome coronavirus (MERS-CoV), and it is hypothesized that a “silencing” of pain through subversion of VEGF-A/NRP-1 signaling may underlie increased disease transmission in asymptomatic individuals [29].
There are three other regions of interest within the spike protein RBD that may also contribute to spike protein bioactivity. The first region is predicted with high probability to be an allergenic sequence [30]. This region could therefore contribute to anaphylaxis in some patients that have received viral vector and/or mRNA COVID-19 vaccines. The second RBD region of interest potentially allows the spike protein to bind to amyloid-forming heparin-binding proteins, which could lead to accelerated aggregation of amyloid proteins within the brain [31]. This supports Classen’s concern that COVID-19 vaccines could potentially induce prion disease [32]. The third region of interest within the RBD contains seven predicted molecular sites that share similarities to different toxins or virulence factors from 12 different bacterial species, 2 malarial parasites and influenza A [33] (Figure 1).
There is one final aspect of this spike protein that warrants consideration regarding its bioactivity, and this stems from the hypothesis that COVID-19-associated multi-system inflammatory syndrome in children (MIS-C) and the cytokine storm observed in adult patients with severe COVID-19 is mediated by spike protein superantigenic activity. Rivas et al. [34] have built on this hypothesis, first by drawing parallels between these two COVID-19 conditions and toxic shock syndrome (TSS). The superantigen Staphylococcus Enterotoxin B (SEB), associated with TSS, is a biotoxin that causes polyclonal T-cell activation and proliferation, which leads to massive production of pro-inflammatory cytokines. These researchers used structure-based computer modelling to discover an SEB-like sequence (glutamic acid661–arginine685) near the spike protein S1/S2 cleavage site that exhibits high binding affinity to both the T-cell receptor (TCR) β chain and co-stimulatory molecule CD28 [35] (Figure 1). They also identified several neurotoxin-like sequences within the spike protein; one (threonine299–tyrosine351) also displayed a high tendency to bind to the TCR, and another is an ICAM1-like region (aparagine280–threonine286) that is predicted to stabilize interactions between the spike protein and the TCR. These researchers also demonstrated TCRV/β skewing of the T cell response in COVID-19 patients with more severe and hyper-inflammatory clinical courses, which is consistent with spike protein superantigen activity. Additionally, they showed that the SARS-CoV-2 mutation aspartic acid839–tyrosine predictably enhanced binding affinity of the spike protein to the TCR, and later this group also provided evidence that a repurposed anti-SEB antibody could prevent SARS-CoV-2 infection in vitro [36].
Collectively, the diverse bioactivity of the SARS-CoV-2 spike protein makes this an ideal target for the immune system to neutralize the virus, and all the current COVID-19 vaccine platforms have focused on this spike protein because it is highly immunogenic [37]. However, we should also be cognizant of these bioactive properties when designing COVID-19 vaccines to ensure that only nontoxic immunogenic portions of the spike protein are expressed, and that their expression is both temporally and spatially limited and does not provide selection pressure driving viral mutation. The mRNA vaccines have been designed to allow a host cell to express the spike protein in its cell membrane [38], and the expression of the spike protein throughout the body is dependent on the biodistribution of LNPs—which primarily relocate to the spleen and liver, but have also been found in various other tissues [39][40]. We currently have no idea how long spike proteins are expressed by different host cells and in what tissues spike protein expression can occur because biodistribution studies on the spike protein have not been carried out to date [40]. The mRNA sequence has also been modified by manufacturers, with the addition of proline residues at positions 986 and 987, which could allow them to reside longer in the plasma membrane [39]. A recent pre-print by Patterson et al. [41] indicated that a subset of monocytes from COVID-19 patients contained SARS-CoV-2 S1 mRNA and proteins for as long as 15 months post-acute infection. This raises the possibility of the spike protein being expressed by maternal immune cells in colostrum and milk from COVID-19 positive mothers; thus, the biodistribution of spike mRNA and protein could be especially relevant during lactation. A recent study by Golan et al. [42] suggested that biodistribution of mRNA to milk during lactation is not a concern, as none was detected in milk from 6 mothers 4–48 h post-Pfizer–BioNTech and Moderna vaccination. However, a study demonstrated that following COVID-19 mRNA vaccination, exosomes expressing spike protein could be detected in plasma up to 4 months post-vaccination [43], which is concerning because we, and others [44][45], have shown that exosomes can be shed in bodily fluids such as colostrum and milk.
2. The SARS-CoV-2 Spike Protein Triggers Autoimmune Responses
Autoimmune diseases can be triggered by viral infections and some vaccines, and are more common to females [46]. There is mounting evidence to support the hypothesis that SARS-CoV-2 infection is a risk factor for autoimmune disease in predisposed individuals [47][48][49][50]. Autoimmune diseases manifest as hyper-stimulated immune responses against autoantigens, which are normally tolerated by the immune system. The proposed mechanisms of autoimmune response during SARS-CoV-2 infection have been previously discussed [50][51] and include molecular mimicry, bystander activation, epitope spreading, and polyclonal lymphocyte activation by SARS-CoV-2 superantigens. Molecular mimicry describes structural similarities between SARS-CoV-2 antigens and autoantigens that are recognized by immune cells (i.e., cytotoxic T cells) and immunoglobulins (i.e., autoantibodies and antiphospholipid antibodies) in cross-reactive epitopes. When autoantigens are targeted by these effectors, this can lead to immune-mediated tissue damage, and if autoreactive memory B-cells and T-cells are generated, this can lead to chronic disease. Bystander activation involves immune-mediated tissue damage resulting from a nonspecific and over-reactive antiviral innate immune response, such as the cytokine storm that has been described in severely impacted COVID-19 patients. In this case, tissue and cellular components become exposed during damage, and are then ingested by phagocytic cells and presented as autoantigens to autoreactive T helper and cytotoxic T cells, which contribute to ongoing immune-mediated pathology. Epitope spreading refers to ongoing sensitization to autoantigens as the disease progresses, which can lead to progressive and chronic disease. A recent study by Zuo et al. [52] implicated anti-NET antibodies as potential contributors of COVID-19 thromboinflammation; NETs are neutrophil extracellular traps that are produced by hyperactive neutrophils that have either come into contact with SARS-CoV-2 or have been activated by platelets and prothrombotic antibodies. These NETs are cytotoxic to pulmonary endothelial cells, and Zuo et al. discovered that anti-NET antibodies contribute to NET stabilization, which may impair their clearance and exacerbate thromboinflammation. Very recently, NETs were also implicated in VIPIT following the Oxford/AstraZeneca vaccine [53], but the potential involvement of anti-NET antibodies remains to be determined.
Autoimmune diseases can be triggered by viral infections and some vaccines, and are more common to females [3]. There is mounting evidence to support the hypothesis that SARS-CoV-2 infection is a risk factor for autoimmune disease in predisposed individuals [94,95,96,97]. Autoimmune diseases manifest as hyper-stimulated immune responses against autoantigens, which are normally tolerated by the immune system. The proposed mechanisms of autoimmune response during SARS-CoV-2 infection have been previously discussed [97,98] and include molecular mimicry, bystander activation, epitope spreading, and polyclonal lymphocyte activation by SARS-CoV-2 superantigens. Molecular mimicry describes structural similarities between SARS-CoV-2 antigens and autoantigens that are recognized by immune cells (i.e., cytotoxic T cells) and immunoglobulins (i.e., autoantibodies and antiphospholipid antibodies) in cross-reactive epitopes. When autoantigens are targeted by these effectors, this can lead to immune-mediated tissue damage, and if autoreactive memory B-cells and T-cells are generated, this can lead to chronic disease. Bystander activation involves immune-mediated tissue damage resulting from a nonspecific and over-reactive antiviral innate immune response, such as the cytokine storm that has been described in severely impacted COVID-19 patients. In this case, tissue and cellular components become exposed during damage, and are then ingested by phagocytic cells and presented as autoantigens to autoreactive T helper and cytotoxic T cells, which contribute to ongoing immune-mediated pathology. Epitope spreading refers to ongoing sensitization to autoantigens as the disease progresses, which can lead to progressive and chronic disease. A recent study by Zuo et al. [99] implicated anti-NET antibodies as potential contributors of COVID-19 thromboinflammation; NETs are neutrophil extracellular traps that are produced by hyperactive neutrophils that have either come into contact with SARS-CoV-2 or have been activated by platelets and prothrombotic antibodies. These NETs are cytotoxic to pulmonary endothelial cells, and Zuo et al. discovered that anti-NET antibodies contribute to NET stabilization, which may impair their clearance and exacerbate thromboinflammation. Very recently, NETs were also implicated in VIPIT following the Oxford/AstraZeneca vaccine [100], but the potential involvement of anti-NET antibodies remains to be determined.
SARS-CoV-2 spike protein superantigen activity was discussed earlier. Superantigens are known to trigger the cytokine storm that can lead to immune-mediated multiple organ dysfunction syndrome, and this is often followed by immune suppression that can lead to persistent infection [101]. Superantigens such as SEB have been shown to exacerbate autoimmune disorders (i.e., experimental autoimmune encephalomyelitis and experimental multiple sclerosis) in mice models [102]. Recently, Jacobs proposed that long-COVID could be due in part to SARS-CoV-2 superantigen-mediated immune suppression, leading to persistent systemic SARS-CoV-2 infection [98]. In terms of pregnancy, prenatal exposure of rats to SEB was shown to attenuate the development and function of regulatory T cells in adult offspring [103] and alter the behaviour (i.e., increased anxiety and locomotion) of mice offspring [104].
Among the proposed mechanisms contributing to autoimmune responses during COVID-19, molecular mimicry has recently taken the front stage. A number of studies have found homologies between SARS-CoV-2 amino acid and human protein amino acid residues [105,106], and more specifically, between the spike protein and human proteins [107,108,109]. Additionally, some of these cross-reactive regions were immunogenic epitopes, meaning that they can bind to MHC I or II molecules on antigen-presenting cells, thereby activating autoreactive B and T cells that elicit an autoimmune response. Martínez et al. [108] for example, identified common host-like motifs in the SARS-CoV-1 and SARS-CoV-2 spike proteins nested in B and T cell epitopes. Morsy and Morsy also identified SARS-CoV-2 spike protein epitopes for MHC I and II molecules that were cross-reactive with the homeobox protein 2.1 (NKX2-1) and ATP-binding cassette sub-family A member 3 (ABCA3) lung proteins [109]. Kanduc and Shoenfeld searched for overlapping SARS-CoV-2 spike protein hexa- and hepta-peptides across mammalian proteomes and found a large number of matches within the human proteome; these authors stated that this is evidence of molecular mimicry, contributing to SARS-CoV-2-associated diseases [107]. Dotan et al. [110] also recently identified 41 immunogenic penta-peptides within the SARS-CoV-2 spike protein that are shared with 27 human proteins related to oogenesis, placentation and/or decidualization, implicating molecular mimicry as a potential contributor to female infertility. Vojdani and Kharrazian also demonstrated that anti-SARS-CoV-2 human IgG monoclonal antibodies cross-reacted with 28 out of 55 human tissue antigens derived from various tissues (i.e., mucosal and blood-brain barrier, thyroid, central nervous system, muscle and connective tissue), and BLAST searches revealed similarities and homologies between the SARS-CoV-2 spike protein and human proteins [111]. In terms of the COVID-19 vaccines, molecular mimicry has also been implicated in myocarditis, an AVR associated with the COVID-19 mRNA vaccines [112]. Huynh et al. [113] also recently identified autoantibodies as the potential cause of VIPIT; these autoantibodies were found to bind to PF4 and allowed for Fc receptor-mediated activation of platelets, which could initiate coagulation, leading to thrombocytopenia and thrombosis. These findings have raised concerns over the possibility that anti-SARS-CoV-2 spike protein antibodies may be responsible for VIPIT. Greinacher A et al. [114] investigated this hypothesis and found that SARS-CoV-2 spike protein and PF4 share at least one similar epitope. However, when they used purified anti-PF4 antibodies from patients with VIPIT, none of the anti-PF4 antibodies cross-reacted with SARS-CoV-2 spike protein. They, therefore, concluded that the vaccine-induced immune response against the SARS-CoV-2 spike protein was not the trigger causing VIPIT.
Anti-idiotypic antibodies were also proposed as an autoimmune response following SARS-CoV-2 infection [121]. In this study, Arthur et al. detected ACE2 autoantibodies in convalescent plasma from previously infected patients, which were also correlated with anti-spike protein RBD antibody levels. Since patients with ACE2 autoantibodies also had less plasma ACE2 activity, these authors hypothesized that the ACE2 autoantibodies were anti-idiotypic antibodies that could interfere with ACE2 function and contribute to post-acute sequelae of SARC-CoV-2 infection (PASC, or “long-COVID”). We are unaware of ACE2 autoantibody levels being assessed following COVID-19 vaccination, so this warrants further investigation.
Collectively, the above autoimmune responses triggered by infection with SARS-CoV-2 or the COVID-19 vaccines suggest potential negative outcomes on fetal and neonatal development, and this should be explored in future studies. As with APS, cytokine storms and thromboinflammation are of concern—as is the potential for autoantibody responses that could target fetal/neonatal proteins.
SARS-CoV-2 spike protein superantigen activity was discussed earlier. Superantigens are known to trigger the cytokine storm that can lead to immune-mediated multiple organ dysfunction syndrome, and this is often followed by immune suppression that can lead to persistent infection [54]. Superantigens such as SEB have been shown to exacerbate autoimmune disorders (i.e., experimental autoimmune encephalomyelitis and experimental multiple sclerosis) in mice models [55]. Recently, Jacobs proposed that long-COVID could be due in part to SARS-CoV-2 superantigen-mediated immune suppression, leading to persistent systemic SARS-CoV-2 infection [51]. In terms of pregnancy, prenatal exposure of rats to SEB was shown to attenuate the development and function of regulatory T cells in adult offspring [56] and alter the behaviour (i.e., increased anxiety and locomotion) of mice offspring [57].
Among the proposed mechanisms contributing to autoimmune responses during COVID-19, molecular mimicry has recently taken the front stage. A number of studies have found homologies between SARS-CoV-2 amino acid and human protein amino acid residues [58][59], and more specifically, between the spike protein and human proteins [60][61][62]. Additionally, some of these cross-reactive regions were immunogenic epitopes, meaning that they can bind to MHC I or II molecules on antigen-presenting cells, thereby activating autoreactive B and T cells that elicit an autoimmune response. Martínez et al. [61] for example, identified common host-like motifs in the SARS-CoV-1 and SARS-CoV-2 spike proteins nested in B and T cell epitopes. Morsy and Morsy also identified SARS-CoV-2 spike protein epitopes for MHC I and II molecules that were cross-reactive with the homeobox protein 2.1 (NKX2-1) and ATP-binding cassette sub-family A member 3 (ABCA3) lung proteins [62]. Kanduc and Shoenfeld searched for overlapping SARS-CoV-2 spike protein hexa- and hepta-peptides across mammalian proteomes and found a large number of matches within the human proteome; these authors stated that this is evidence of molecular mimicry, contributing to SARS-CoV-2-associated diseases [60]. Dotan et al. [63] also recently identified 41 immunogenic penta-peptides within the SARS-CoV-2 spike protein that are shared with 27 human proteins related to oogenesis, placentation and/or decidualization, implicating molecular mimicry as a potential contributor to female infertility. Vojdani and Kharrazian also demonstrated that anti-SARS-CoV-2 human IgG monoclonal antibodies cross-reacted with 28 out of 55 human tissue antigens derived from various tissues (i.e., mucosal and blood-brain barrier, thyroid, central nervous system, muscle and connective tissue), and BLAST searches revealed similarities and homologies between the SARS-CoV-2 spike protein and human proteins [64]. In terms of the COVID-19 vaccines, molecular mimicry has also been implicated in myocarditis, an AVR associated with the COVID-19 mRNA vaccines [65]. Huynh et al. [66] also recently identified autoantibodies as the potential cause of VIPIT; these autoantibodies were found to bind to PF4 and allowed for Fc receptor-mediated activation of platelets, which could initiate coagulation, leading to thrombocytopenia and thrombosis. These findings have raised concerns over the possibility that anti-SARS-CoV-2 spike protein antibodies may be responsible for VIPIT. Greinacher A et al. [67] investigated this hypothesis and found that SARS-CoV-2 spike protein and PF4 share at least one similar epitope. However, when they used purified anti-PF4 antibodies from patients with VIPIT, none of the anti-PF4 antibodies cross-reacted with SARS-CoV-2 spike protein. They, therefore, concluded that the vaccine-induced immune response against the SARS-CoV-2 spike protein was not the trigger causing VIPIT.
3. The SARS-CoV-2 Spike Protein and Antibody-Dependent Enhancement
Anti-idiotypic antibodies were also proposed as an autoimmune response following SARS-CoV-2 infection [68]. In this study, Arthur et al. detected ACE2 autoantibodies in convalescent plasma from previously infected patients, which were also correlated with anti-spike protein RBD antibody levels. Since patients with ACE2 autoantibodies also had less plasma ACE2 activity, these authors hypothesized that the ACE2 autoantibodies were anti-idiotypic antibodies that could interfere with ACE2 function and contribute to post-acute sequelae of SARC-CoV-2 infection (PASC, or “long-COVID”). We are unaware of ACE2 autoantibody levels being assessed following COVID-19 vaccination, so this warrants further investigation.
Collectively, the above autoimmune responses triggered by infection with SARS-CoV-2 or the COVID-19 vaccines suggest potential negative outcomes on fetal and neonatal development, and this should be explored in future studies. As with APS, cytokine storms and thromboinflammation are of concern—as is the potential for autoantibody responses that could target fetal/neonatal proteins.
While antibodies have a number of important effector activities against SARS-CoV-2, including limiting viral attachment to epithelial cells and viral neutralization, non-neutralizing antibodies that enhance viral entry into host cells can sometimes also be generated; this immunological phenomenon is referred to as antibody-dependent enhancement (ADE). Since the early days of the COVID-19 pandemic, concerns have been raised about the possibility of ADE occurring, as it has been reported that both SARS-CoV-1 and MERS-CoV infect various animal models via ADE [122,123]. Ricke [123] proposed that SARS-CoV-2 may leverage Fc receptors for host cell invasion, and this may contribute to cytokine storms, leading to adult multi-system inflammatory syndrome, and also infant MIS-C—the latter presumably being mediated by passive transfer of maternal anti-SARS-CoV-2 antibodies that have become bound to Fc receptors on infant mast cells or macrophages [124]. While the potential risk of this type of ADE occurring in response to COVID-19 vaccines remains unknown, experience with SARS-CoV-1 spike protein vaccines demonstrates that it is indeed a possibility which warrants further investigation [123].
A second type of ADE involves non-neutralizing antibodies binding to and then eliciting conformational changes to viral proteins that can lead to enhanced viral adhesion to host cells [122]. Liu et al. [125] recently screened a panel of anti-SARS-CoV-2 spike protein monoclonal antibodies derived from COVID-19 patients and found that some of these antibodies that bind to the N-terminal domain of the spike protein induce open confirmation of the RBD, which enhances the binding capacity of the spike protein to ACE2 and the infectivity of SARS-CoV-2. Interestingly, these infection-enhancing antibodies have been identified in both uninfected and infected blood donors and have been detected at high levels in severe COVID-19 patients; their presence in uninfected people implies that these individuals may be at risk of severe COVID-19 if they later become infected with SARS-CoV-2 [125]. Another recent study has suggested that people may be at risk of infection by the SARS-CoV-2 Delta variant if they were vaccinated against the Wuhan strain spike sequence because the Delta variant is well-recognized by infection-enhancing antibodies targeting the N-terminal domain of the spike protein [126].
A number of murine studies have demonstrated that non-neutralizing maternal antibodies can increase the risk of neonatal disease. For example, pregnant mice infected with different strains of Dengue virus (DENV) display maternal anti-DENV IgG that is passively transferred during gestation and enhances the severity of offspring disease (i.e., hepatocyte vacuolation, vascular leakage, lymphopenia and thrombocytopenia) following infection with the heterotypic strain [127], and breastfeeding has been shown to extend the window of ADE [128]. In terms of anti-SARS-CoV-2 antibodies, the passive transfer of anti-SARS-CoV-2 neutralizing antibodies has been detected in milk samples collected from women with COVID-19 [129]; however, non-neutralizing antibodies were not assessed. Anti-spike protein antibodies (IgG and IgA) have also been detected in milk samples from lactating mothers who were vaccinated with SARS-CoV-2 mRNA vaccines [130]; however, their neutralization/non-neutralization status was not assessed. Therefore, we have no data to determine whether or not a passive transfer of ADE can occur during SARS-CoV-2 infection or COVID-19 vaccination, and so this warrants further investigation.
3. The SARS-CoV-2 Spike Protein and Antibody-Dependent Enhancement
While antibodies have a number of important effector activities against SARS-CoV-2, including limiting viral attachment to epithelial cells and viral neutralization, non-neutralizing antibodies that enhance viral entry into host cells can sometimes also be generated; this immunological phenomenon is referred to as antibody-dependent enhancement (ADE). Since the early days of the COVID-19 pandemic, concerns have been raised about the possibility of ADE occurring, as it has been reported that both SARS-CoV-1 and MERS-CoV infect various animal models via ADE [69][70]. Ricke [70] proposed that SARS-CoV-2 may leverage Fc receptors for host cell invasion, and this may contribute to cytokine storms, leading to adult multi-system inflammatory syndrome, and also infant MIS-C—the latter presumably being mediated by passive transfer of maternal anti-SARS-CoV-2 antibodies that have become bound to Fc receptors on infant mast cells or macrophages [71]. While the potential risk of this type of ADE occurring in response to COVID-19 vaccines remains unknown, experience with SARS-CoV-1 spike protein vaccines demonstrates that it is indeed a possibility which warrants further investigation [70].
A second type of ADE involves non-neutralizing antibodies binding to and then eliciting conformational changes to viral proteins that can lead to enhanced viral adhesion to host cells [69]. Liu et al. [72] recently screened a panel of anti-SARS-CoV-2 spike protein monoclonal antibodies derived from COVID-19 patients and found that some of these antibodies that bind to the N-terminal domain of the spike protein induce open confirmation of the RBD, which enhances the binding capacity of the spike protein to ACE2 and the infectivity of SARS-CoV-2. Interestingly, these infection-enhancing antibodies have been identified in both uninfected and infected blood donors and have been detected at high levels in severe COVID-19 patients; their presence in uninfected people implies that these individuals may be at risk of severe COVID-19 if they later become infected with SARS-CoV-2 [72]. Another recent study has suggested that people may be at risk of infection by the SARS-CoV-2 Delta variant if they were vaccinated against the Wuhan strain spike sequence because the Delta variant is well-recognized by infection-enhancing antibodies targeting the N-terminal domain of the spike protein [73].
A number of murine studies have demonstrated that non-neutralizing maternal antibodies can increase the risk of neonatal disease. For example, pregnant mice infected with different strains of Dengue virus (DENV) display maternal anti-DENV IgG that is passively transferred during gestation and enhances the severity of offspring disease (i.e., hepatocyte vacuolation, vascular leakage, lymphopenia and thrombocytopenia) following infection with the heterotypic strain [74], and breastfeeding has been shown to extend the window of ADE [75]. In terms of anti-SARS-CoV-2 antibodies, the passive transfer of anti-SARS-CoV-2 neutralizing antibodies has been detected in milk samples collected from women with COVID-19 [76]; however, non-neutralizing antibodies were not assessed. Anti-spike protein antibodies (IgG and IgA) have also been detected in milk samples from lactating mothers who were vaccinated with SARS-CoV-2 mRNA vaccines [77]; however, their neutralization/non-neutralization status was not assessed. Therefore, we have no data to determine whether or not a passive transfer of ADE can occur during SARS-CoV-2 infection or COVID-19 vaccination, and so this warrants further investigation.