2. Activation of CO2 on Heterogeneous Catalyst Surface
The interaction of CO
2 molecules with metal-based surfaces has attracted intense research attention for a long time. Reduction of CO
2 by H
2 has been studied on some transition metals (e.g., Cu, Co, and Ni) and metal oxide catalysts (e.g., TiO
2, CeO
2, and In
2O
3). The catalytic surfaces for the activation of CO
2 include metal sites, metal-oxide interfaces, and oxygen vacancies. For facile CO
2 activation and conversion, efforts should be made to know, design, and optimize functional sites in heterogeneous catalysts, which involves constructing surface sites with a charge density gradient capable of reorganizing the electronic structures of CO
2 and polarizing the adsorbed species
[11][85].
Metal-based catalysts are capable of effectively activating CO
2 even in the absence of hydrogen
[12][27]. These include pure metal surfaces, doped or promoted metal surfaces, and supported metal nanoparticles. On supported metal surfaces, CO
2 activation occurs mainly through acceptance of charge from the metal; the oxide support plays a big part in the activation process through different mechanisms such as acid sites interactions and provision of oxygen vacant sites
[13][86]. The metal surface is also important in the dissociation of H
2 in CO
2 hydrogenation reactions
[3][6].
2.1. CO2 Activation on Representative Pure Metals
Metal nanoparticles sit on a metal oxide serve as active sites for the electron transfer. Transition metals such as Cu, Ni, and Fe are particularly energetic for activating CO
2. Their surfaces possess high binding abilities to CO
2. DFT calculations evidenced that on Pt, Rh, Ni, Cu, Ag, and Pd (111) surfaces, the affinity toward oxygen is different for the metals, which selects their reaction pathways for the CO
2 activation in the RWGS) reaction. Pt, Ag, and Pd tend to favor the COOH-mediated mechanism, whereas Rh, Ni, and Cu dissociate CO
2 into CO and O
[14][87]. This difference underlines the variation in the CO
2 dissociation barrier of the different metal groups. Thus, the nature of the interaction between the adsorbed O and the surface is critical for determining the CO
2 dissociation barrier. In the activation of CO
2 via the charge transfer mode, which is prevalent on metal surfaces, the partial and full charge transfer leads to the formation of CO
2δ− and CO
2−, respectively. The degree of charge transfer can be analyzed on metal surfaces after adsorption. Physisorption and chemisorption of CO
2 on single crystal surfaces of various metals have been studied by means of DFT calculations
[15][88]. Chemisorption states are highly dependent on both the metal itself and adsorption sites. Chemisorbed CO
2 molecules often have a bent structure with O–C–O angle varying between 121 and 140° compared with the nearly linear coordination of the physisorbed CO
2 molecules, and their extent depends on the kind of metal surface. In other words, the degree of CO
2 activation varies with metal surfaces. Similar behavior can be observed for the amount of charge transferred. According to Wang et al.
[16][89], who investigated CO
2 chemisorption on nine transition metal surfaces (Fe, Co, Ni, Cu, Rh, Pd, Ag, Pt, and Au), the adsorption strength is affected by both the
d-band center of the metal surface and the charge transfer, which control the activation of the C=O bond. It is possible to promote chemisorption by adjusting the properties of the catalysts, such as the catalyst surface area, surface defects, basic sites, and the addition of promoters
[17][31].
The coordination of CO
2 with metals can take different modes via the electron-deficient C atom as the electron acceptor and the C=O bonds or O atoms as the electron donor. Electron-rich metal surfaces such as Ni (110) and Cu (100) generally activate CO
2 by attaching to the carbon atom
[18][19][90,91]. In the process, electrons in the d
z2 orbital of the metal transfer to the unpopulated antibonding π* orbital of the CO
2 molecule. Consequently, negatively charged surfaces are efficient for CO
2 activation via the charge transfer mode. Such charged surfaces can be generated through inserting strong metal-support interaction, bimetallic and ligand effects, etc.
[11][85]. Electron-deficient metal surfaces, such as Ti, Cr, V, and Mn, favor the end-on coordination with CO
2 [20][92]. In this mode, the bending or distortion of the linear CO
2 molecule is difficult
[11][85]. For metal centers with combined binding behavior, i.e., possessing both an electron acceptor site and an electron donor site, CO
2 adsorption generally prefers the bridge sites. More efficient activation can result through this mechanism and lead to bending of the linear CO
2 molecule from different sites
[11][85].
Investigations by DFT calculations revealed the characteristic adsorption and activation of CO
2 on Rh, Pd, Pt, Ni, Fe, Cu, Re, Al, Mg, and Ag metals
[14][21][87,93]. Strong evidence has been provided for the formation of CO
2− according to spectroscopic results. Depending on the type of metal, CO
2 can also dissociate into CO and O or be transformed into CO
32− and CO. The activation of CO
2 on Pt, Rh, Ni, Cu, Ag, and Pd followed different elementary steps as a result of the different levels of interaction of the metals with adsorbed oxygen in the RWGS reaction. Metals with high affinities toward oxygen presented lower activation barriers, leading to facile hydrogenation reactions
[14][87]. The presence of preadsorbed oxygen was responsible for forming carbonates of different structures
[14][21][87,93]. On most metal surfaces, CO
2 activation is highly surface orientated, pressure- and particle size-dependent
[16][22][23][24][89,94,95,96]. For e.g., Yu et al.
[24][96] demonstrated through spin-polarized DFT calculations that adsorption and dissociation of CO
2 were dependent on the Co particle size. They showed that Co
55 nanoclusters had the highest CO
2 dissociation activity in comparison to Co
13 and Co
38. However, Co
13 activated CO
2 with the smallest O–C–O angle (123°) against 137° for both Co
38 and Co
55 nanoclusters.
Cu-based materials have gained much attention in the CO
2 conversion process due to their wide applicability in the different conversion processes and low cost
[9][25][12,97]. Despite these and other massive studies, the activation of CO
2 on Cu catalysts is still an issue due to the poor understanding of its mechanism. Studies have shown that CO
2 interacts weakly on low-index Cu surfaces under UHV conditions
[26][27][98,99]. However, a recent study found Cu (100) surface to be more active in dissociating CO
2 than Cu (111), producing atom oxygen
[19][91]. Ambient pressure X-ray photoelectron spectroscopy (APXPS) and DFT calculations revealed the activation of CO
2 on Cu surfaces. APXPS showed that CO
2 adsorbed as CO
2δ− on Cu (111) surface under a pressure of 0.01 mbar at 300 K. With an increase in pressure to 1 mbar, adsorbed CO
2δ− partially transformed into carbonate as a result of the disproportionation reaction between CO
2 molecules. Subsequent annealing at 400 K or higher temperatures led to the dissociation of CO
2δ− and carbonate and the formation of a chemisorbed oxygen-covered surface. On Cu (110) surface, the CO
2δ− gradually dissociated into CO and chemisorbed oxygen under the same CO
2 pressure at room temperature. On both surfaces, atomic oxygen was generated that catalyzed the self-deactivation of CO
2 adsorption. The DFT results, which collaborated experimental findings, further indicated that the Cu (110) surface was more active than the Cu (111) surface in breaking C–O bonds
[23][95]. Comparing the adsorption of CO
2 on Cu (111), (100), and (110) surfaces, it was found that CO
2 molecules aligned parallel to Cu (111) and (100), whereas a vertical configuration was more stable for the adsorbed CO
2 on Cu (110) with one of two oxygen atoms towards the surface. The degree of CO
2 activation followed the order: Cu (110) > Cu (100) > Cu (111)
[16][89]. A decrease in the activation energies for CO
2 dissociation has been observed when Cu surface
s have step or kink defects in comparison with the flat surface. Chemisorption of CO
2 was reported on Cu stepped surfaces
[26][27][28][29][98,99,100,101].
Ni-based catalysts can dissociate and convert CO
2 into value-added products, such as methane; thus, a fundamental understanding of the interaction between CO
2 and Ni surface at the atomic level is crucial to design even more efficient Ni-based catalysts. According to theoretical studies, the surface orientation of Ni influences the activation of CO
2 by altering the energetics for subsequent C–O bond cleavage
[30][102]. Ab initio calculations using slab models have shown that CO
2 reactions on model Ni are surface sensitive, with reactivity in the following trend: Ni (110) > Ni (100) > Ni (111)
[30][31][102,103]. Experimental investigations revealed the capability of Ni (110) surface to molecularly adsorb and subsequently dissociate CO
2 at room temperature
[32][104]. By using in situ APXPS, carbonate was identified as the dominating surface intermediate at room temperature upon CO
2 adsorption on Ni (111) and Ni (110) surfaces
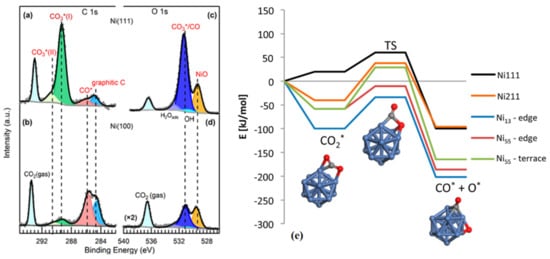
[33][34][35][105,106,107]. However, where there are multiple dissociation products, their distribution depends on the surfaces. Carbonate, CO, and graphitic carbon were all observed on both Ni (111) and Ni (100) surfaces under a CO
2 pressure of 0.2 Torr. Ni (111) was predominantly covered with carbonate, whereas adsorbed CO* and graphitic carbon were prevalent on the Ni (100) surface as indicated in
Figure 1a–d
[35][107]. The CO
2 adsorption and dissociation on ideal Ni (111) and stepped Ni (211) surfaces are shown in
Figure 1e. It can be seen that CO
2 adsorption is endothermic by 20 kJ·mol
−1 on Ni (111) surface, whereas it is exothermic by 40 kJ·mol
−1 on Ni (211) surface
[36][108].
Figure 1. Ambient pressure XPS spectra of (
a,
b) C 1s, and (
c,
d) O 1s Ni (111) and Ni (100), respectively. CO
2 adsorption was performed under 0.2 Torr CO
2 at room temperature. Reproduced with permission from
[35][107]. Copyright 2019 American Chemical Society; (
e) Energy profile diagram for the CO
2 activation on Ni (111) and Ni (211) surfaces, edges of Ni
13 particle, and edges and terraces of the Ni
55 particle and the activation geometries on the Ni
13. Reproduced with permission from
[36][108]. Copyright 2016 American Chemical Society.
Based on the experimental results of the ultrahigh vacuum (UHV), Auger electron spectroscopy (AES), temperature-programmed desorption (TPD), and high-resolution electron energy loss spectroscopy (HREELS), it is difficult for CO
2 to adsorb on a clean Pt (111) surface between 110 and 300 K, but the dissociation capability can be improved by doping alkali metals such as potassium
[37][38][109,110]. Conversely, it was reported that Pt foil treated with CO
2 in the gas phase during in situ UHVXPS experiments at 77 K formed chemisorbed CO species
[39][111]. In collaboration with the latter assertion, on the clean Pt (111) surface, CO
2 dissociated into adsorbed CO and O at both room and elevated temperatures
[40][112]. The production of adsorbed CO increased upon the introduction of H
2 (hydrogenation reaction). This observation was obvious for the CO
2 adsorption at all temperatures and led to the deoxygenation (consumption of oxygen) of the surface, cleaning the sites for further CO production and desorption from the surface at elevated temperatures. The Pt surface was active in the RWGS reaction. At low pressure, the RWGS was initiated at 300 °C; on the contrary, at high pressure (H
2:CO
2 of 150 mtorr: 15 mtorr), a low temperature (200 °C) favored the initiation of the RWGS reaction, and the conversion of CO
2 increased with increasing temperatures
[40][112]. Moreover, under a pressure of 40 mtorr of pure CO
2 and at temperatures below 150 °C, graphitic carbon has been also observed as a product of the Boudouard reaction. IR spectroscopy revealed the size-dependent nature of CO
2 adsorption on Pt clusters. On small Pt clusters anions (Pt
n−, n = 4–7), CO
2 was highly activated but remained molecularly adsorbed on Pt
4−. On large clusters, dissociative adsorption was observed
[10][13].
2.2. CO2 Activation on Bimetallic/Alloyed Catalyst Surfaces
Bimetallic surfaces are highly unique and active for a wide range of CO
2 transformation reactions due to the electronic and geometric alterations within the structure. These alterations could be observed as a change in the morphology of metal, adsorption mode and configuration, and chemical ordering with varying composition and particle size
[5][41][8,34]. This can help to control the adsorption properties to attain desired adsorption coverages. Numerous studies have discussed both activation of CO
2 and the subsequent conversion of CO
2 to fuels and chemicals on bimetallic catalysts. Ko et al.
[15][88] studied the CO
2 activation and adsorption by performing DFT calculations on a range of bimetallic alloy surfaces. The dissociation energy barriers of CO
2 were screened by combining Bronsted–Evans–Polanyi (BEP) relation, scaling relation, and surface mixing rule. It was found that CO
2 dissociated into CO and O, which sum of their adsorption energies was linearly related to both the energy for CO
2 dissociation and that for CO
2δ− adsorption. The activation of CO
2 proceeded through a direct dissociation (CO
2 → CO + O) mechanism in three successive elementary steps: physisorption of CO
2 from the gas phase on the metal surface, chemisorption of CO
2δ− from the physisorbed CO
2, and direct dissociation of CO
2δ− into CO and O.
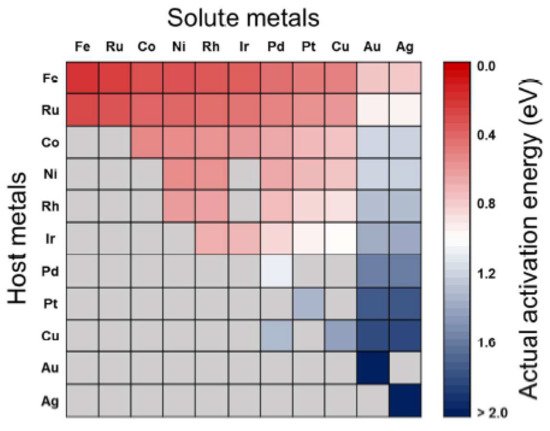
The predicted activation energies for the CO
2 dissociation on bimetallic alloy surfaces are shown in
Figure 2, with the row and column indicating the host and solute metals of the bimetallic alloys, respectively. The alloying effect would lead to a reduction in surface reactions when the solute metals are placed in the bulk region. In the figure, the activation energy (
Eaact) decreases from right to left and from bottom to top, thus alloys with relatively low activation energies (~0.75 eV) are Fe-, Ru-, Co-, Ni-, Rh-, and Ir-based alloys, whereas Pd-, Pt-, and Cu-based alloys possess high activation energies (~1.51 eV). Still, some metals, including Ru-, Co-, Ni-, Rh-, Ir-, and Cu-based alloys, have activation energies in between the extremes (0.76–1.50 eV). This picture makes sense for fabricating bimetallic catalysts for CO
2 conversion reactions by manipulating the activation energies
[15][88].
Figure 2. Screening for
Eaact for CO
2 dissociation on pure metals and bimetallic alloys. Gray cells indicate the bimetallic alloys which are not preferred to the surface segregation of solute atoms.
Eaact were estimated by combining BEP relation, scaling relation, and surface mixing rule. Reproduced with permission from the authors of
[15][88]. Copyright 2016 American Chemical Society.
The mechanisms of CO
2 dissociation on bimetallic clusters (Pt
3Ni
1, Pt
2Ni
2, and Pt
1Ni
3) were investigated and characterized by the activation barrier
[42][21]. Compared with the single metallic clusters (Pt
4 and Ni
4), results showed the activation barrier to be between those of the monometallic clusters. Increasing Ni atoms in the bimetallic clusters moderately raised the activation barrier, which means that such a bimetal combination could have a significant negative impact on the activity of Pt for CO
2 reaction
[42][21].
Results from an ab initio study of chemisorption and activation of CO
2 on Pt-based transition-metal nanoalloys on 55-atom nanoclusters (Pt
nTM
55−n), where n = 0, 13, 42, 55, and TM = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Au, indicated a linear correlation between the interaction energy, charge transfer from the nanoclusters toward CO
2 and the bent CO
2 angle
[5][8]. It was further realized that the interaction energy was enhanced for larger angles and molecular charge. With 55 atoms for Cu, Ag and Au in the Pt alloy, a change from physisorption to chemisorption was observed, whereas the strong interaction energy of CO
2 with Os
55, Ru
55, and Fe
55 can be decreased by alloying with Pt
[5][8]. It means that certain metals (Fe, Co, and Ni) activate CO
2 more strongly as monometals than in an alloyed form due to weaker adsorption energy in the latter.
2.3. CO2 Adsorption and Activation on Metal Oxide Surfaces
2.3.1. Metal Oxide
Various metal oxides (MO
x) or (M
xO
y) are investigated as supports or as catalysts for CO
2 conversion, including In
2O
3, CeO
2, ZnO, ZrO
2, TiO
2, and CeO
2. Metal oxide surfaces consist of both metal (M
n+) and oxygen (O
2−) ions, which are effective sites for CO
2 activation. The activation can occur by coordination to one or two adjacent metal sites through the terminal oxygen atoms or C atom of the CO
2 molecule, forming monodentate or bidentate carbonate species
[43][114]. Interaction of the C atom is on the surface oxygen sites of the metal oxide. The CO
2 activation can also occur via the σ-bond and π-bonds activation on metal ions and oxygen ions, respectively, as observed upon chemisorption on metal oxides applied as catalyst supports
[44][115]. Due to the large surface areas, switchable redox properties, and rich oxygen vacancies, metal oxides can act as adsorption and activation sites for small molecules, including O
2, H
2, and CO
2 [45][116]. The surface oxygen vacancies interact with the carbon and/or oxygen atoms of CO
2 through which electron transfer from the oxide defective site to adsorbed CO
2 becomes feasible. One example is In
2O
3, which is rich in oxygen vacancies and has shown a high activity for CO
2 activation and methanol synthesis by hydrogenation
[45][116].
On oxide surfaces, generally, CO
2 interactions can vary from physisorption to chemisorption, the extent that affects the structure and reactivity of the adsorbed CO
2, and the kinetics and mechanisms of surface catalytic reactions
[43][114]. The surface structure is important for CO
2 adsorption and activation
[46][117]; thus, the interaction of CO
2 with metal oxides can be structure-dependent. It was found that CO
2 adsorption on Zn
2GeO
4 (001) was higher than that on Zn
2GeO
4 (010) surface
[46][117]. The interaction with (010) surface led to bidentate carbonate species, whereas on the (001) surface, stronger interaction with CO
2 resulted in a bridged carbonate-like species. The strongest adsorption based on calculated CO
2 adsorption energies was around the surface oxygen vacancy site on both surfaces. Analysis of the LDOS and Mulliken charge for adsorbed CO
2 on perfect Zn
2GeO
4 surfaces revealed that CO
2 formed a CO
2δ− species upon accepting electrons from the surface. CO
2 molecule was found to be activated on the CuO surfaces ((011), (111), and (−111)), with strong adsorption only on the (011) surface. The CuO (111) and CuO (−111) surfaces showed relatively weak adsorption. CO
2 activation was characterized by structural transformations and charge transfer that resulted in the formation of bent CO
2δ− species with an elongation of the C–O bonds
[22][94].
The ability of metal oxides to bind and activate CO
2 depends greatly on several factors, including their preparation methods, physiochemical properties, redox properties, and electronic and geometric structures
[36][43][47][108,114,118]. The preparation methods were found to impact the properties of CeO
2 nanostructures for the photocatalytic reduction of CO
2 [48][119]. A high surface area was obtained for catalysts synthesized through the sunlight-assisted combustion process, in addition to possessing a small particle size, high concentration of oxygen vacancies, and a narrow bandgap. Compared to that prepared from the conventional combustion process with a spongy-like structure, a porous network consisting of small and uniformed pores was also obtained for the sunlight-assisted process. The superior catalytic properties could be attributed to the novel properties endowed by solar irradiation during the synthesis process. As demonstrated with the CeO
x/Cu catalyst, the important roles of metal oxide ions were revealed on the catalytic cycle of H
2O and CO
2 activation. The Cu phase was reduced into Cu
0 that promoted Ce
4+ reduction into Ce
3+. H
2O and CO
2 activation occurred on the Ce
3+ sites. Without the presence of Cu, Ce
3+ would lead to oxidation into Ce
4+. However, in contact with Ce
4+, Cu
0 reacted to form Cu
+ and Ce
3+, sustaining ceria in the more active state. The cycle is closed when Cu
+ reduced to Cu
0 [49][120]. This synergistic effect afforded the catalyst with high reactivity in the RWGS reactions. The chemisorption of CO
2 molecules on CeO
2 at RT as studied using in situ DRIFTS indicated adsorption at both the Ce
3+ and Ce
4+ sites, although adsorption was also found at the oxygen sites that resulted in carbonates and bicarbonates species
[50][121]. In the same study, the CO
2 chemisorption on TiO
2 under similar reaction conditions and instrumentation was observed at both Ti
3+ and Ti
4+ sites, exhibiting O–C–O vibrations at 1667 and 1248 cm
−1 and 2339–2345 cm
−1, respectively
[50][121]. The CO
2 molecules adsorbed at Ti
3+ sites formed CO
2− species, which concentration increased with the amount of oxygen vacancies present. Like with CeO
2, CO
2 chemisorption at the oxygen sites formed carbonates and bicarbonates species. It was observed that the interaction between TiO
2 and CO
2 molecules is somewhat weak compared with that of CeO
2. Such weak interactions can be improved by doping TiO
2 with CeO
2. CeO
2 doping can improve the interaction of TiO
2 with CO
2 as a result of the introduction of Ce
3+, which strengthens the bonding of CO
2 with catalyst surfaces and enhances the production of bidentate carbon species that can readily be transformed to surface CO
2− in the presence of H
2O under solar irradiation. The formation of adsorbed species of CO
2 over CeO
2/TiO
2 could be attributed to the binding of CO
2 species to Ti/Ce atoms that have reductive capabilities under photo-irradiation. Furthermore, the Ce
3+ availability from CeO
2 facilitates photogenerated charge separation; thus, the CO
2 adsorption and enhanced charge separation can be tuned for increased activity of CeO
2/TiO
2 catalyst
[51][122]. The surface area of materials positively correlates with their adsorption capacity. It was found that the Bi
12O
17Cl
2 nanotubes had a higher adsorption capacity for CO
2 (~4.3 times) than bulk Bi
12O
17Cl
2 due to the higher BET specific surface area of the former. As a result, the effective adsorption of CO
2 on Bi
12O
17Cl
2 nanotubes over bulk Bi
12O
17Cl
2 favored the photocatalytic process
[52][123]. In addition, the high surface area correlated with strong adsorption. Weak chemisorption of CO
2 has been reported for CeO
2 nanostructures with low exposed surface area
[50][121]. Mesostructured photocatalyst displayed improved activity for CO
2 reduction into CH
4 due to the presence of high specific surface area and well-developed mesostructure that enhanced adsorption of CO
2 [53][124]. Highly mesoporous In(OH)
3 synthesized via the sol-gel hydrothermal treatment exhibited ~20-fold higher efficiency for CO
2 reduction in comparison with those lacking mesopores
[54][125]. It is reported that the methanol activity of the In
2O
3 catalyst could also be improved by increasing the (111) surface area
[55][126].
2.3.2. Characteristic Adsorption of Representative Metal Oxides
Ceria (CeO
2) has shown catalytic activity in the reduction of CO
2 to liquid fuels and chemicals. It has rich oxygen vacancies and high oxygen storage/release capacity. Several studies demonstrating the interaction of CO
2 with high-surface-area ceria catalysts have been reported. As noted in ref
[56][127], CO
2 dissociates into CO and an oxygen-containing surface species on the surface Ce
3+ ions, which are considered active sites for CO
2 activation due to the formation of carbonates or inorganic carboxylates. Graciani et al. reported a highly active CeO
x/Cu nanoparticles catalyst for methanol synthesis from CO
2 [57][128]. The catalyst activated CO
2 as CO
2δ− and exhibited a faster methanol production rate than Cu/ZnO, on which CO
2 was chemisorbed as CO
32−. A study on the CO
2 adsorption sites of CeO
2 (110) surface using DFT was carried out by Cheng et al.
[58][129]. Reduced and stoichiometric ceria (110) surfaces were compared. Results revealed that CO
2 adsorption on the reduced ceria (110) surface was thermodynamically favored than on the stoichiometric ceria (110) surface. Furthermore, the most stable adsorption configuration consisted of CO
2 adsorbed parallel to the reduced ceria (110) surface at the oxygen vacancy. Upon adsorption, the CO
2 molecule distorted out of the plane and formed carbonates with the remaining oxygen anion at the surface
[58][129]. It was suggested that the structural changes in the catalyst after CO
2 adsorption were due to charge transfer between the surface and adsorbate molecule. The formation of two different adsorbate species: a carbonate and a weakly bound and linear physisorbed species, were observed upon exposure of reduced CeO
2−x (110) substrates to CO
2 at low temperatures. There was no evidence for the formation of CO
2δ−. Furthermore, based on angle-dependent C K-edge NEXAFS spectra, the most preferred orientation of the adsorbate could not be observed. The physisorbed CO
2 species and carbonate species were completely desorbed at 250 and 500 K, respectively. The authors remarked that it is most unlikely that the activation of CO
2 on the reduced CeO
2−x (110) surface was via breaking the C=O bond to form CO and O. However, on fully oxidized CeO
2 (110), CO
2 adsorbed as a carbonate which was completely decomposed and desorbed as CO
2 at 400 K
[59][130]. CO
2 adsorbed on the CeO
2 (111) surface formed monodentate carbonate species found to be most stable on CeO
2 at low coverages
[60][131]. Increasing the CO
2 coverage destabilized the formed species, indicating a mixed adsorption mechanism with both carbonate and linearly adsorbed CO
2 species. Although CeO
2 has been studied for CO
2 reduction reactions, the insights into CO
2 adsorption, activation, and reaction on ceria surfaces are not yet fully understood.
Titania (TiO
2) possesses good photocatalytic properties for many chemical reactions, including CO
2 reduction. Since its first demonstration in the photoelectrochemical CO
2 reduction to formic acid and formaldehyde by Inoue et al.
[61][132], TiO
2-based materials have attracted great research interests in CO
2 photoreduction reactions. The adsorption properties of CO
2 on both the rutile and anatase phases of TiO
2 have been widely studied using various surface science techniques
[62][63][64][65][133,134,135,136]. Sorescu et al.
[66][137] investigated the adsorption and dissociation of CO
2 on an oxidized anatase (101) surface using dispersion-corrected DFT and found CO
2 to adsorbed at a fivefold coordinated Ti site in a tilted configuration. Based on in situ FTIR experiments, the CO
2 adsorption formed CO
32− and CO
2 bonded to Ti, with absorption bands at 1319, 1376, 1462, 1532, 1579, and 2361 cm
−1. The band at 2361 cm
−1 was assigned to adsorbed CO
2 with Ti–O–C–O adsorption configuration
[67][138]. The 1319 and 1579 cm
−1 bands were assigned to bidentate carbonate, while the band at 1461 cm
−1 was due to monodentate or free carbonate. Under the vacuum condition, the intensities of all of the bands were reduced at 35 °C. The bidentate carbonate was the predominant species for CO
2 on TiO
2.
TheThe scanning tunneling microscopy (STM) scanning tunne
ling microscopy (STM) ennabled a study of the dissociation of CO
2 adsorbed at the oxygen vacancy of TiO
2 (110) at the single-molecule level
[68][139]. It was found that the electrons injected from the STM tip into the adsorbed CO
2 caused its dissociation into CO and O, and the released O was observed to heal the oxygen vacancy. According to experimental analysis, ~1.4 eV above the conduction band minimum of TiO
2 is needed for the electron induction process to dissociate CO
2. The formation of a transient negative ion by the injected electron is an important step in the CO
2 dissociation, and this can only be possible above the threshold voltage. TiO
2 modified with metal oxide nanoclusters possess enhanced activity to adsorb and convert CO
2 [69][70][26,140]. The Bi
2O
3-TiO
2 heterostructures obtained by modifying TiO
2 with Bi had low coordinated Bi sites in the nanoclusters and a valence band edge consisting mainly of Bi–O states due to the presence of the Bi lone pair. Upon interaction of CO
2 with the reduced heterostructures, CO or CO
2− were observed mainly through electron transfer to CO
2, and the Bi
2O
3–TiO
2 heterostructures became oxidized in the process with adsorbed CO
2 in carbonate form
[70][140]. In a related study, clean or hydroxylated extended rutile and anatase TiO
2 surfaces modified with Cr nanoclusters presented an upshift valence band edge related to the existence of Cr 3d–O 2p interactions, which promoted the CO
2 activation.
[69][26]. The activated CO
2 molecule reduced its O–C–O angle to 127–132° and increased the C–O bond length to 1.30 Å. It was concluded that the strong CO
2–Cr–O interaction induced the structural distortions.
Iron oxides (FeO
x) are an important component of catalysts for the conversion of CO
2 to hydrocarbons (liquid fuels). The adsorption and activation of CO
2 on FeO
x have been investigated by researchers
[71][72][73][141,142,143]. It is suggested that Fe
2+ and Fe
3+ cations are crucial for CO
2 adsorption. Using TPD, Pavelec et al.
[71][141] observed a weak interaction between CO
2 and Fe
3O
4 (001) surface. On this surface, CO
2 molecules existed in the physisorbed state as they desorbed at a low temperature (115 K). However, strong CO
2 adsorption was observed on the defects and surface Fe
3+ sites. Weak CO
2 adsorption has also been observed on Fe
3O
4 (111) as investigated by various experimental techniques
[74][144]. At different CO
2 dosages and temperatures (between 120 and 140 K), TPD experiments suggested CO
2 adsorb very weakly on a regular Fe
3O
4 (111) surface. However, CO
2 chemisorption was also observed but at relatively long CO
2 exposure times
[74][75][144,145] via binding to under-coordinated oxygen sites
[75][145]. The formation of chemisorbed species such as carboxylates and carbonates was facilitated by surface imperfections. Conclusively, FeO
x exhibit weak interaction with CO
2 molecules, and studies are recommended in this direction to adjust its CO
2 adsorption strength.
ZrO
2 has been demonstrated as catalyst support for the CO
2 hydrogenation reactions to a variety of products. In a study on CO
2 hydrogenation on Cu/ZrO
2 catalyst using the first-principles kinetic Monte Carlo simulations by Tang et al.
[76][146], the authors showed that CO
2 prefers to adsorb on the bare ZrO
2 nanoparticles surface rather than at the Cu/ZrO
2 interface. This led to the bending of the CO
2 molecule with a calculated adsorption energy of 0.69 eV. The stretching of the C–O bonds and charge transfer from the ZrO
2 surface to the antibonding 2π
μ orbital of CO
2 were also observed. On the bare ZrO
2 surface, bidentate bicarbonate (HCO
3) was formed upon CO
2 adsorption based on observable IR frequencies at ~1225, ~1620, and ~3615 cm
−1 [77][147].
3. Conclusion
CO2 is a kinetically stable molecule that requires high energy input for the C–O bond breaking. Its proper activation can reduce the high energy barrier substantially, easing conversion by various processes. The CO2 activation is an important step that precedes the conversion of CO2 to chemicals and fuels. It can be effected in the presence of a catalyst by altering the CO2 electronic and molecular properties. Upon accepting an extra electron from a substrate, the neutral CO2 molecule forms an anion with a full charge (CO2–) or partial charge (CO2δ–). Some metals and metal oxides are efficient catalysts for CO2 conversion reactions; thus, they should be good for CO2 activation. In general, metal nanoparticles serve as active sites for electron transfer, with certain factors such as change in morphology of metal particles, nanoparticle size, adsorption mode and configuration, and chemical ordering as the CO2 activation marker. The interaction of CO2 with some pure metals is rather weak but can be improved by incorporating promoters (e.g., alkali metals) with low electronegativity. Metal oxide nanoparticles are utilized as supports or as catalysts for CO2 conversion. Their surfaces comprise both metal (Mn+) and oxygen (O2–) ions, which can act as active sites for CO2 activation. They can activate CO2 by coordinating to one or two adjacent metal sites through the terminal oxygen atoms of the CO2 or by interaction of the carbon atom of CO2 with surface oxygen sites. A particularly interesting feature in metal oxides is the oxygen vacancies that facilitate CO2 adsorption and activation.
CO2 is a kinetically stable molecule that requires high energy input for the C–O bond breaking. Its proper activation can reduce the high energy barrier substantially, easing conversion by various processes. The CO2 activation is an important step that precedes the conversion of CO2 to chemicals and fuels. It can be effected in the presence of a catalyst by altering the CO2 electronic and molecular properties. Upon accepting an extra electron from a substrate, the neutral CO2 molecule forms an anion with a full charge (CO2–) or partial charge (CO2δ–). Some metals and metal oxides are efficient catalysts for CO2 conversion reactions; thus, they should be good for CO2 activation. In general, metal nanoparticles serve as active sites for electron transfer, with certain factors such as change in morphology of metal particles, nanoparticle size, adsorption mode and configuration, and chemical ordering as the CO2 activation marker. The interaction of CO2 with some pure metals is rather weak but can be improved by incorporating promoters (e.g., alkali metals) with low electronegativity. Metal oxide nanoparticles are utilized as supports or as catalysts for CO2 conversion. Their surfaces comprise both metal (Mn+) and oxygen (O2–) ions, which can act as active sites for CO2 activation. They can activate CO2 by coordinating to one or two adjacent metal sites through the terminal oxygen atoms of the CO2 or by interaction of the carbon atom of CO2 with surface oxygen sites. A particularly interesting feature in metal oxides is the oxygen vacancies that facilitate CO2 adsorption and activation.