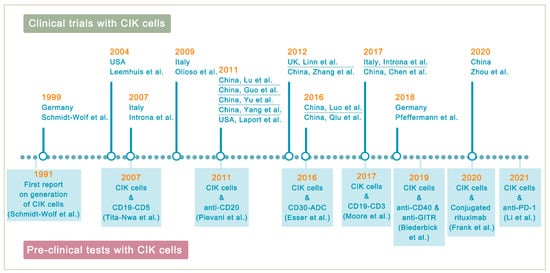
Lymphoma is a heterogeneous group of neoplasms including over 70 different subtypes. Its biological characteristic of deriving from lymphoid tissues makes it ideal for immunotherapy. Cytokine-induced killer (CIK) cells therapy is one of the adoptive immunotherapy which eliminates cancer cells by transfusing immune cells that have been expanded and activated in vitro. CIK cells are heterogeneous in nature and composed of CD3+CD56- (T cells), CD3-CD56+ (NK cells), and CD3+CD56+ (NKT cells) cell populations. The CD3+CD56+ subset is considered as the major effector cells that exert potent anti-tumor cytotoxicity in a major histocompatibility complex (MHC)-unrestricted manner. From 1991 to 2020, there are nearly 20 clinical trials conducted for the treatment of lymphoma. Moreover, a number of pre-clinical approaches are being investigated to improve CIK cell therapy to enhance its anti-tumor activity.
- lymphoma
- cytokine-induced killer cells
- adoptive cell transfer
- immunotherapy
1. Introduction
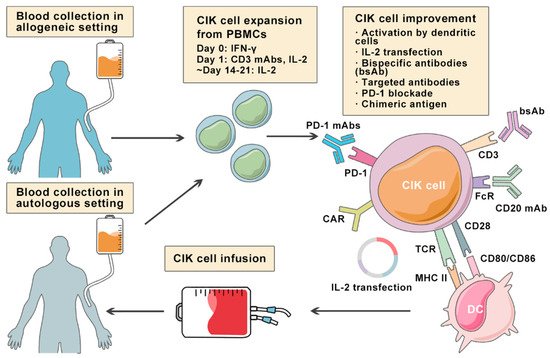
2. Cytokine-Induced Killer Cells
| CIK Cells | CAR-T Cells | BiTEs | |||
|---|---|---|---|---|---|
| Manufacturing Process | PBMCs sequentially activated with 1000 IU/m IFN-γ on day 0, and 50 ng/mL anti-CD3 mAb and 600 IU/mL IL-2 on the following day; IL-2 supplemented every 2–3 days | T cells with CAR gene transduction primarily by lentiviruses; CARs consist of an scFv based ectodomain for antigen-binding, a transmembrane domain, and an endodomain containing TCR CD3ζ chain with or without costimulatory signaling components | Antibodies designed to bind to a selected TAA and CD3 on T cells simultaneously; produced in bioreactors by mammalian cell lines as secreted polypeptides | ||
| Effector Cells | CD3+CD56+ cells | Mostly αβ-TCR+ T cells | Endogenous CD8+ or CD4+ T cells | ||
| Cell Source | Autologous/allogeneic | Autologous/allogeneic | Autologous | ||
| Target Antigen | MIC A/B and ULBP1–4 | CD19, CD20, CD22, CD30, BCMA, etc. | CD19 | ||
| MHC Restriction | Dual-functional capability (non-MHC-restricted and TCR-mediated lysis) | TAA recognition by CARs is MHC-unrestricted | MHC-unrestricted | ||
| Mechanism | Release of perforin and granzyme B from CIK cells | Release of perforin and granzyme B from CAR-T cells | Activating T cells to release perforin and granzyme B by linking TAAs to CD3 on T cells | ||
| Toxicities and Side effects | Low-grade toxicities including fever, chills, fatigue, headache, and skin rash; grade 3 and 4 toxicities are rare; limited GVHD response in the allogeneic setting | CRS, ICANS, and MAS/HLH; potentially life-threatening | CRS and ICANS; severe toxicities are one of the major concerns | ||
| Clinical Efficacy | Varied due to heterogeneity of expension method and study design | Axicabtagene ciloleucel: 83% ORR, 58% CR; Tisagenlecleucel: 54% ORR, 40% CR; Lisocabtagene maraleucel: 73% ORR, 53% CR | [13][14][15] | Blinatumomab: 36% to 69% ORR in relapsed/refractory NHL | [16][17] |
3. Improving CIK Cell Therapy on Lymphoma
References
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin lymphoma. Lancet 2017, 390, 298–310.
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390.
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA A Cancer J. Clin. 2017, 68, 116–132.
- Introna, M. CIK as therapeutic agents against tumors. J. Autoimmun. 2017, 85, 32–44.
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991, 174, 139–149.
- Verneris, M.R.; Karami, M.; Baker, J.; Jayaswal, A.; Negrin, R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood 2004, 103, 3065–3072.
- Karimi, M.; Cao, T.M.; Baker, J.A.; Verneris, M.R.; Soares, L.; Negrin, R.S. Silencing Human NKG2D, DAP10, and DAP12 Reduces Cytotoxicity of Activated CD8+T Cells and NK Cells. J. Immunol. 2005, 175, 7819–7828.
- Frigault, M.J.; Maus, M.V. State of the art in CAR T cell therapy for CD19+ B cell malignancies. J. Clin. Investig. 2020, 130, 1586–1594.
- Goebeler, M.-E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434.
- Diefenbach, A.; Jamieson, A.; Liu, S.D.; Shastri, N.; Raulet, D. Ligands for the murine NKG2D receptor: Expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 2000, 1, 119–126.
- Ramos, C.A.; Heslop, H.E.; Brenner, M.K. CAR-T Cell Therapy for Lymphoma. Annu. Rev. Med. 2016, 67, 165–183.
- Zhang, Y.; Schmidt-Wolf, I.G.H. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J. Cell. Physiol. 2020, 235, 9291–93033.
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42.
- Abramson, M.J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, D.M.A.; Wang, M.L.; Arnason, J.E.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, M.C.; et al. Pivotal Safety and Efficacy Results from Transcend NHL 001, a Multicenter Phase 1 Study of Lisocabtagene Maraleucel (liso-cel) in Relapsed/Refractory (R/R) Large B Cell Lymphomas. Blood 2019, 134, 241.
- Bachanova, V.; Westin, J.; Tam, C.; Borchmann, P.; Jaeger, U.; McGuirk, J.; Holte, H.; Waller, E.; Jaglowski, S.; Bishop, M.; et al. Correlative Analyses of Cytokine Release Syndrome and Neurological Events in Tisagenlecleucel-Treated Relapsed/Refractory Diffuse Large B-Cell Lymphoma Patients. Clin. Lymphoma Myeloma Leuk. 2019, 19, S251–S252.
- Viardot, A.; Goebeler, M.-E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416.
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients with Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results from a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111.
- Ansell, S.M. Fundamentals of immunology for understanding immunotherapy for lymphoma. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 585–589.
- Tita-Nwa, F.; Moldenhauer, G.; Herbst, M.; Kleist, C.; Ho, A.D.; Kornacker, M. Cytokine-induced killer cells targeted by the novel bispecific antibody CD19xCD5 (HD37xT5.16) efficiently lyse B-lymphoma cells. Cancer Immunol. Immunother. 2007, 56, 1911–1920.
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Ap-plication of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551.
- Circosta, P.; Elia, A.R.; Landra, I.; Machiorlatti, R.; Todaro, M.; Aliberti, S.; Brusa, D.; Deaglio, S.; Chiaretti, S.; Bruna, R.; et al. Tailoring CD19xCD3-DART exposure enhances T-cells to eradication of B-cell neoplasms. OncoImmunology 2018, 7, e1341032.
- Pievani, A.; Belussi, C.; Klein, C.; Rambaldi, A.; Golay, J.T.; Introna, M. Enhanced killing of human B-cell lymphoma targets by combined use of cytokine-induced killer cell (CIK) cultures and anti-CD20 antibodies. Blood 2011, 117, 510–518.
- Esser, L.; Weiher, H.; Schmidt-Wolf, I. Increased Efficacy of Brentuximab Vedotin (SGN-35) in Combination with Cytokine-Induced Killer Cells in Lymphoma. Int. J. Mol. Sci. 2016, 17, 1056.
- Frank, M.J.; Olsson, N.; Huang, A.; Tang, S.-W.; Negrin, R.S.; Elias, J.E.; Meyer, E.H. A novel antibody-cell conjugation method to enhance and characterize cytokine-induced killer cells. Cytotherapy 2020, 22, 135–143.
- Biederbick, K.D.; Schmidt-Wolf, I.G. Efficacy of cytokine-induced killer cells targeting CD40 and GITR. Oncol. Lett. 2019, 17, 2425–2430.
- Li, Y.; Sharma, A.; Bloemendal, M.; Schmidt-Wolf, R.; Kornek, M.; Schmidt-Wolf, I.G.H. PD-1 blockade enhances cytokine-induced killer cell-mediated cytotoxicity in B-cell non-Hodgkin lymphoma cell lines. Oncol. Lett. 2021, 22, 613.