Deferasirox is a first-line therapy for iron overload that can sometimes cause kidney damage. A proximal tubulopathy pattern may be observed on treatment with deferasirox. Since deferasirox-associated kidney damage is dose-dependent, physicians should prescribe the lowest efficacious dose.
1. Introduction
Iron overload secondary to regular blood transfusions may result in injury and dysfunction of the heart, liver, anterior pituitary, pancreas, and joints. Both parenteral iron chelation with deferoxamine and oral chelation with deferiprone or deferasirox have been shown to reduce iron overload and organ damage
[1][2][1,2]. Due to its efficacy and ease of use, deferasirox 20–30 mg/kg once-daily is currently the first-line therapy for iron overload secondary to blood transfusions
[1][2][1,2]. It has been known for about 10 years that an increase in circulating creatinine occurs in about one out of ten cases
[3]. This tendency is most likely to occur in well-chelated patients with circulating ferritin 1000 µg/L or less
[4].
2. Kidney Tubular Damage Secondary to Deferasirox
The majority (61%) of the 57 patients were ≤18 years of age (Table 1). Three-quarters of the cases were affected by a thalassemia syndrome. Laboratory features consistent with kidney damage were mostly observed >6 months after starting a standard dose deferasirox therapy, although this information was not available in more than half of the cases. A recurrence of the kidney damage was noted in nine of the 18 patients, who were again exposed to deferasirox (usually in a reduced dose).
Table 1. Characteristics of 57 patients 3 to 78, median 15 years of age with kidney damage on deferasirox therapy. Data are presented as median [with interquartile range] or as frequency (with percentage).
| Female, N (%) |
27 |
47 |
| Male, N (%) |
30 |
53 |
| Age |
|
|
| Years, median [interquartile range] |
15 [6.7–21] |
| ≤18 years, N (%) |
35 |
61 |
| Underlying transfusion-dependent disease |
|
|
| Thalassemia syndrome, N (%) |
46 |
81 |
| Diamond Blackfan anemia, N (%) |
5 |
8.8 |
| Allogenic stem cell transplantation, N (%) |
3 |
5.3 |
| Other conditions ◆, N (%) |
3 |
5.3 |
| Deferasirox dose☩ |
|
|
| 20–30 mg/kg/day, N (%) |
46 |
87 |
| 31–42 mg/kg/day, N (%) |
7 |
13 |
| Duration of deferasirox therapy ✙ |
|
|
| ≤1 month, N (%) |
5 |
20 |
| 2–6 months, N (%) |
4 |
16 |
| >6 months, N (%) |
16 |
64 |
| Time to recovery after therapy withdrawal * |
|
|
| Information not given, N (%) |
37 |
65 |
| ≤1 week, N (%) |
2 |
3.5 |
| 2–4 weeks, N (%) |
3 |
5.3 |
| >6 months, N (%) |
5 |
8.8 |
| Persistent abnormalities reported |
2 |
3.5 |
| Deferasirox therapy rechallenge, N (%) |
18 |
32 |
| Relapse of kidney damage, N (%) |
9 |
16 |
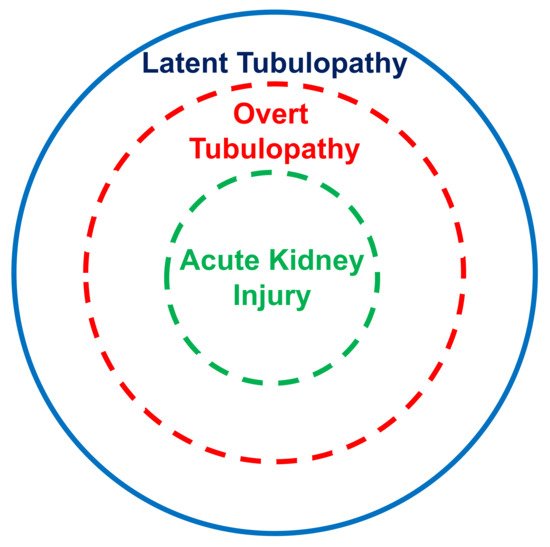
The kidney tubular damage associated with oral deferasirox therapy may present in three ways: (a) abnormal urinary findings consistent with latent tubular damage; (b) overt acid–base or electrolyte abnormalities; and (c) acute kidney injury (always associated with abnormal urinary findings and with an electrolyte or acid–base imbalance).
The kidney tubular damage caused by deferasirox characteristically presents with renal glucosuria, excessive ß
2-microglobulin excretion, generalized aminoaciduria, non-gap metabolic acidosis, hypophosphatemia, and hypouricemia. Hence, these data suggest the existence of a proximal tubular disturbance. Proximal tubular defects are typically observed on treatment with aminoglycosides, nucleotide reverse transcriptase inhibitors, or platinum compounds and result from mitochondrial toxicity
[5][6][30,31]. No kidney damage was so far reported on treatment with other iron chelators such as deferiprone or deferoxamine
[2][7][2,32]. These clinical observations are supported by in vitro studies: it was found that deferasirox, but not other chelators, induces a dramatic swelling of mitochondria and decreases the cellular ATP content
[8][33]. Iron being essential for kidney tubular cells, it is currently assumed that deferasirox associated tubular damage results from excessive chelation of iron within these cells, which is likely superior to that observed with other chelators
[9][34]. Furthermore, the kidney damage caused by deferasirox is likely dose-dependent
[9][34]. Finally, pharmacogenetics might be a relevant determinant of deferasirox toxicity
[9][10][34,35].
In this analysis, most cases with acid–base or electrolyte abnormalities simultaneously presented with urinary abnormalities. Furthermore, all cases with kidney injury concurrently presented with an acid–base or electrolyte imbalance and with urinary abnormalities. It is therefore tempting to assume that the latent tubulopathy represents the early stage, the overt tubulopathy the middle stage, and finally the manifest acute kidney injury the advanced stage of damage induced by deferasirox (Figure 12). This hypothesis deserves confirmation in longitudinal studies.
Figure 12. Suggested stratification of the kidney damage associated with deferasirox therapy.
It has been speculated but not proven that iron overload may per se induce kidney damage
[1][2][1,2]. Furthermore, sickle cell disease and perhaps also some other transfusion-dependent conditions may cause kidney abnormalities
[11][36].
3. Conclusions
Aminoglycoside-class antimicrobials, nucleotide reverse transcriptase inhibitors, and platinum compounds occasionally cause a dose- and time-related kidney disease. Similar damage may be observed on treatment with the iron-chelating agent deferasirox but not with deferoxamine or deferiprone. Deferasirox-associated kidney damage is dose-dependent and more likely to occur when iron stores are low. It has therefore been recommended
[4][9][4,34] that physicians prescribe the lowest possible dose to achieve a satisfactory iron burden and consider discontinuing therapy if circulating ferritin is 1000 μg/L or less.