Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Ruth Birner-Grünberger.
Hepatic stellate cells (HSC) are the major cellular drivers of liver fibrosis. Upon liver inflammation caused by a broad range of insults including non-alcoholic fatty liver, HSC transform from a quiescent into a proliferating, fibrotic phenotype. Here we report proteomic changes during activation of HSC in the serum activated HSC model LX-2.
- hepatic stellate cells
- activation
- fibrosis
1. Introduction
Liver fibrosis is the replacement of parenchymal liver tissue by non-parenchymal scar tissue [1]. This process mostly occurs due to liver inflammation from alcohol abuse [2], viral infection [3], or a non-alcoholic fatty liver [4]. In early stages, liver fibrosis is reversible upon treatment of the underlying etiology [5]. If left untreated liver fibrosis can progress to more severe conditions like cirrhosis and liver cancer [6]. However, there is still no approved treatment for the progression of fibrosis other than the removal of the source of inflammation [7].
Most fibrotic tissue generation can be attributed to a specialized cell type in the liver, the hepatic stellate cells (HSC) [8]. Under healthy liver conditions, these non-parenchymal cells merely act as a storage buffer for retinol (vitamin A), which is deposited as retinylesters in cytosolic lipid droplets (LD) [9]. Upon sustained liver inflammation, HSC transform from a quiescent (resting) phenotype towards a myofibroblast-like phenotype [1]. This transformation, termed “activation”, is a key event in liver fibrosis [10]. HSC display distinct characteristic changes during activation, which allows them to proliferate at an increased rate [11], migrate [12] towards the site of liver damage, lose their LD and increase their production of fibrotic proteins [13]. If we can improve our understanding of the cellular and molecular processes involved in HSC activation, we might identify targets to revert HSC to their quiescent phenotype, therefore alleviating fibrosis in a damaged liver.
Immortalized human HSC cell lines, which closely reflect primary HSC in vivo, are widely available [14]. These cell lines allow for accessible in-depth mechanistic investigations without the use of cancer cell lines or animal models. Often these HSC cell lines are treated with CCl4 or TGF-β to induce activation in vitro [15]. We chose a well described HSC cell line developed in 2005, termed Lieming Xu 2 (LX-2) [16], which displays a low activation (quiescent) state under low serum conditions [17]. The addition of serum activates LX-2 cells [17[17][18][19],18,19], making it an accessible model for studying HSC activation without the need for CCl4 or TGF-β. It is noteworthy to add that LX-2 cells never achieve a fully quiescent state when grown on plastic dishes [16].
2. Hepatic Stellate Cells Are Activated by Fetal Bovine Serum
The LX-2 cell line shows characteristic features of primary HSC in vivo and displays a quiescent phenotype in growth medium with low serum concentrations but gets activated in high serum concentrations [16]. We here employed this easy and robust approach to study changes between quiescent and activated LX-2 cells.
We treated LX-2 cells with either 1% or 10% FBS for 24 h. Their activation status was confirmed by investigation of α-smooth muscle actin (α-SMA; Figure 1), a protein highly expressed in activated HSC which enhances the contractility of cells [20] and has been prominently featured as a reliable marker for HSC activation [20,21,22][20][21][22].
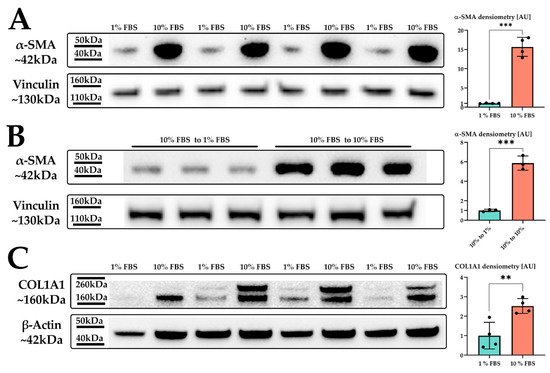
Figure 1. LX-2 HSC activation by serum. (A) α-SMA protein expression of LX-2 cells under low (1% FBS) or high (10% FBS) serum concentration. Vinculin was used as loading control. (B) α-SMA protein expression of LX-2 cells incubated in 10% FBS for 24 h, then for additional 24 h in either 1% FBS (10% FBS to 1 % FBS) or 10% FBS (10% FBS to 10% FBS). Vinculin was used as loading control. (C) COL1A1 protein expression of LX-2 cells under low (1% FBS) or high (10% FBS) serum concentration. One additional band can be seen at ~200 kDa, which was not used for quantification. Loading control was β-actin. ** p < 0.01; *** p < 0.001, unpaired t-test, two-sided.
Indeed, we observed a strong upregulation in α-SMA protein expression upon increased serum concentration in the growth medium (Figure 1A). We next investigated whether the serum activation of LX-2 cells was reversible. For this, we incubated LX-2 cells in 10% FBS containing DMEM for 24 h, replaced the media with 1% FBS containing DMEM for additional 24 h and subsequently analyzed the expression of α-SMA. Figure 1B suggests that LX-2 cells can switch back from their activated to their quiescent state, depending on the serum concentration in the medium. In addition to α-SMA, activated HSC produce increased amounts of extracellular matrix (ECM) proteins, such as collagen [23,24][23][24]. Collagen 1 α1 (COL1A1) is frequently used as a marker for fibrosis [25] and therefore can also be used to indicate the activation status of HSC. Correspondingly, LX-2 cells indeed showed increased deposition of COL1A1 protein upon serum activation for 24 h (Figure 1C).
3. Serum Activated Hepatic Stellate Cells Show a Phenotype Characteristic of Activated Hepatic Stellate Cells
We next analyzed the phenotype of activated LX-2 cells. During activation, HSC transform into a myofibroblast-like phenotype, which is accompanied by an increased growth rate [11], enhanced migration [12], and the loss of LD volume [26]. We saw enhanced growth of LX-2 cells upon an increase in serum concentration after 48 h (Figure 2A).
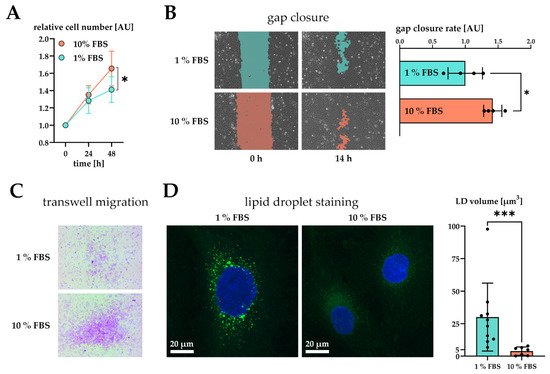
Figure 2. Effect of serum activation on LX-2 cell phenotype. (A) Relative proliferation of LX-2 cells under low (1% FBS) or high (10% FBS) serum concentrations, normalized to 0 h. (B) Left: gap closure assay of LX-2 cells in either 1% FBS or 10% FBS after 0 h and 14 h. Right: Quantification of gap closure rate where the increase of occupied space is measured against time and plotted as slope (normalized to 1 % FBS). Higher values represent faster migration. (C) Transwell migration of LX-2 cells through a membrane in either 1% FBS or 10% FBS after 24 h. These images show the underside of the membrane and therefore only migrated cells stained purple. (D) Left, both images: lipid droplets (BOPIPY, green) and nuclei (DAPI, blue) staining of LX-2 cells shown as maximum intensity Z-projection. Right: Quantification of lipid droplet volume per cell. * p < 0.05; *** p < 0.001, unpaired t-test, two-sided.
Furthermore, we tested the migratory properties of activated LX-2 cells. We first performed a gap closure assay (scratch assay) where LX-2 cells must migrate in order to close a gap between confluent cell batches. To suppress serum-driven proliferation effects, we pre-incubated both conditions in 1% FBS and replaced the growth media by fresh 1% or 10% FBS containing DMEM at timepoint 0 h. We then monitored migration of cells for the period of only 14 h during which no growth difference was observed (Figure 2A). The difference in growth was prominent only after 48 h in culture (Figure 1A). As expected, serum activated LX-2 cells migrated faster, leading to quicker gap closure (Figure 2B). Faster migration was further corroborated by performing a transwell migration assay. For this purpose, LX-2 cells were seeded onto porous transwells at 1% or 10% FBS. Serum activated LX-2 cells showed enhanced migration to the lower side of the membrane after 24 h (Figure 2C).
We also investigated whether serum activation of LX-2 cells was accompanied by loss of LD. In order to induce LD formation, LX-2 were first incubated in 200 µM oleic acid at 1% FBS for 24 h and then treated with either 1% or 10% FBS containing DMEM for additional 48 h. We observed a decline in LD volume in serum activated LX-2 cells (Figure 2D). Furthermore, LD in serum activated LX-2 cells appeared to be smaller in accordance with [27].
Altogether, these findings confirm serum activation of LX-2 cells as an accessible option for studying the activated phenotype of HSC in vitro.
4. Proteomic Analysis of Serum Activated Hepatic Stellate Cells Reveals Changes of Several Key Cellular Pathways
To reveal global changes on protein level induced by HSC activation we examined the proteome of serum activated LX-2 cells. After treatment of LX-2 cells with either 1% or 10% FBS for 48 h, cells were harvested and subjected to shotgun proteomics. As a result, we quantified 4598 proteins across six biological replicates per treatment group (quiescent or activated). The protein list was filtered to keep only those proteins quantified in all six replicates in at least one of the treatment groups. The resulting protein matrix comprised of 3163 proteins was subjected to statistical analysis.
Principal component analysis (PCA) of the proteomics dataset revealed a clear separation between serum activated and quiescent LX-2 cells (Figure 3A). Correspondingly, as shown in the volcano blot in Figure 3B, serum activated LX-2 cells undergo a massive change to their proteome during activation; in numbers: 465 proteins were highly significantly up- or downregulated (two-sample t-test, two-sided, S0 = 0.1, permutation-based false discovery rate (FDR) = 0.05, 250 randomizations). In accordance with the observed phenotypes, activated stellate cells had a higher abundance of proteins involved in migration (e.g., fibronectin 1 FN1, angio-associated migratory cell protein AAMP, Figure 3B, marked in purple), reduced levels of proteins involved in lipid biosynthesis (e.g., FASN, CYP51A1, Figure 3B, marked in red) and a prominent up-regulation of ribosomal proteins (Figure 3B, marked in blue).
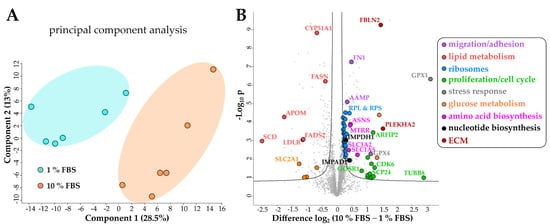
Figure 3. Changes of the LX-2 HSC proteome upon serum activation. (A) Principal component analysis shows a clear separation between serum activated (10% FBS) vs. quiescent (1% FBS) LX-2 cells. (B) Volcano blot of serum activated (10% FBS) vs. quiescent (1% FBS) LX-2 cells at FDR = 0.05 and S0 = 0.1. FDR corrected Student t-test p-value < 0.05, N = 6 biological replicates per group.
4.1. Serum Activated Hepatic Stellate Cells Show Increased Ribosome Biogenesis, Cell Cycle, Cell Migration and Oxidative Stress Related Proteins
To gain an overview of metabolic pathways and protein classes affected by serum activation we used significantly changed proteins as input for protein network and functional enrichment analysis using the STRING [28] database for analysis and Cytoscape [29] for visualization (Figure 4). As one of the most prominently enriched clusters in serum activated LX-2 cells we identified ribosome biogenesis related proteins, including small ribosomal proteins (RPS) as well as large ribosomal subunit proteins (RPL) (gene ontology (GO) process ribosome biogenesis GO:0042254, 49 enriched genes out of 270 pathway genes, FDR: 3.2 × 10−4 for proteins with enrichment values, enrichment score: 1.02665; Figure 4A). Additionally, we identified 32 proteins involved in rRNA processing (GO:0006364), of which 31 were upregulated in serum activated cells.
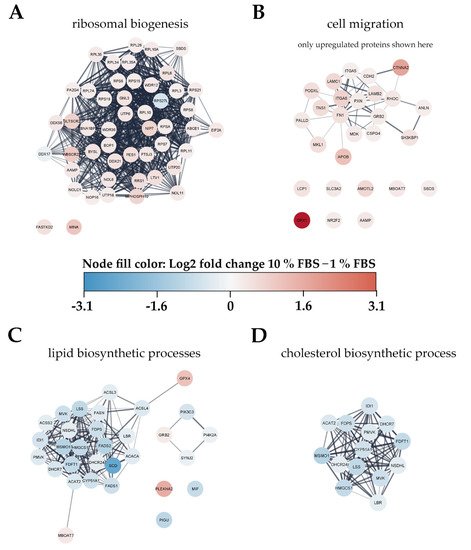
Figure 4. Visualization of changes to the proteome of LX-2 cells during serum activation. The color bar in the center indicates up- (red) or downregulated (blue) proteins in serum activated (10% FBS vs. 1% FBS) LX-2 cells. (A) Ribosomal proteins (small ribosomal subunits RPS and large ribosomal subunits RPL) as well as proteins related to ribosomal biogenesis were upregulated in serum activated LX-2 cells. (B) Many proteins related to cell migration like AAMP and FN1 are upregulated in serum activated LX-2 cells fitting the observed increase in cell migration (Figure 2B,C). (C) Serum activated LX-2 cells show a decreased expression of proteins involved in the lipid biosynthetic process fitting the observed decrease in LD volume (Figure 2D). (D) Supplementary to (C), proteins involved in cholesterol biosynthesis are distinctly downregulated in serum activated LX-2 cells.
Furthermore, we also observed increased expression of Golgi-ER traffic related proteins (Tubulin beta-8 chain TUBB8, Arfaptin-2 ARFIP2, Golgi SNAP receptor complex member 1 GOSR1). These findings are in line with the observation of increased ECM protein production (see Figure 1C).
Additionally, upregulated proteins in serum activated LX-2 cells like cell division protein kinase 6 (CDK6) or kinetochore protein Spc24 (SPC24) imply an upregulation of the cell cycle (Figure 3B, green dots), which can also be seen in Figure 2A where we show increased proliferation in serum activated LX-2 cells. In line with these findings, we found upregulated nucleotide biosynthesis proteins in activated HSC (Figure 3B, black dots).
Interestingly, proteins related to cellular stress response like glutathione peroxidase 1 (GPX1), and GPX4 are highly upregulated in serum activated LX-2 cells (Figure 3B, grey). Glutathione peroxidases were shown to reduce oxidative stress in HSC [30].
Proteomic network analysis also suggested increased expression of proteins related to cell migration, including the aforementioned AAMP and FN1 in serum activated LX-2 cells. Although STRING functional enrichment analysis with values/ranks did not identify cell migration to be significantly enriched, 27 out of 37 matched proteins involved in this GO term were found significantly upregulated. (Figure 4B; cell migration GO process GO:0016477, 37 enriched genes out of 812 pathway genes, FDR: 0.0151 for upregulated proteins only, network strength: 0.35). This observation is in line with the phenotype we describe in chapter 2.1.1 and Figure 2B,C.
4.2. Activated Hepatic Stellate Cells Show a Decrease in Fatty Acid and Cholesterol Biosynthesis
Interestingly, lipid biosynthetic processes were found to be downregulated in serum activated LX-2 cells (GO process GO:0008610, 30 enriched genes out of 585 pathway genes, FDR: 2.8 × 10−4 for proteins with enrichment values, enrichment score: 2.08448), including all fatty acid desaturases (SCD (being the most downregulated protein (6-fold)), FADS1, FADS2), acetyl-CoA-carboxylase 1 (ACACA), and fatty acid synthase (FASN; Figure 4C). Some of these findings are in accordance with previous reports as FASN downregulation was already reported on mRNA level in activated (10 % FBS) as compared to quiescent (2 % FBS) LX-2 cells [18]. These observations are in accordance with the LD volume changes described in chapter 2.1.1 and Figure 2D. However, not only fatty acid de-novo synthesis and desaturation were affected during activation. Cholesterol biosynthesis was another prominently downregulated pathway in activated HSC (GO process GO:0006695, 14 enriched genes out of 41 pathway genes, FDR: 7.4 × 10−3 for proteins with enrichment values, enrichment score: 2.62406). In addition, lipid transport proteins were reduced, e.g., apolipoprotein M (APOM) and low-density lipoprotein receptor (LDLR), see Figure 3B, red dots.
Overall, the proteomic alterations suggest several metabolic changes. Additionally, we identified a downregulation of glucose transporter SLC2A1 in serum activated LX-2 cells, see Figure 3B, yellow dots, suggesting decreased glucose uptake. Lastly, we also detected changes in abundance of several ECM proteins. However, we could not identify upregulation of COL1A1 like we see in our western blot (Figure 1C) or a clear trend in the expression of other collagen proteins in our proteomics dataset. One reason for this might be the proteomics sample preparation: while for western blot analysis trypsin was used for cell harvest, for proteomics analysis physical detachment using cell scrapers was applied. This might suggest that cell scraping does not assure a quantitative collection of collagen proteins. Additionally, crosslinked ECM proteins with low solubility are notoriously difficult to quantify using proteomics [31]. Despite this, we could identify many higher expressed regulators of ECM production, including fibulin-2 (FBLN2) and pleckstrin homology domain-containing family A member 2 (PLEKHA2) in serum activated LX-2 cells (Figure 3B, dark red dots).
References
- Kisseleva, T.; Brenner, D. Molecular and Cellular Mechanisms of Liver Fibrosis and Its Regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166.
- Bataller, R.; Gao, B. Liver Fibrosis in Alcoholic Liver Disease. Semin. Liver Dis. 2015, 35, 146–156.
- Su, T.-H.; Kao, J.-H.; Liu, C.-J. Molecular Mechanism and Treatment of Viral Hepatitis-Related Liver Fibrosis. Int. J. Mol. Sci. 2014, 15, 10578–10604.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922.
- Zoubek, M.E.; Trautwein, C.; Strnad, P. Reversal of Liver Fibrosis: From Fiction to Reality. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 129–141.
- O’Rourke, J.M.; Sagar, V.M.; Shah, T.; Shetty, S. Carcinogenesis on the Background of Liver Fibrosis: Implications for the Management of Hepatocellular Cancer. World J. Gastroenterol. 2018, 24, 4436–4447.
- Altamirano-Barrera, A.; Barranco-Fragoso, B.; Méndez-Sánchez, N. Management Strategies for Liver Fibrosis. Ann. Hepatol. 2017, 16, 48–56.
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42.
- Haaker, M.W.; Vaandrager, A.B.; Helms, J.B. Retinoids in Health and Disease: A Role for Hepatic Stellate Cells in Affecting Retinoid Levels. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158674.
- Reeves, H.L.; Friedman, S.L. Activation of Hepatic Stellate Cells—A Key Issue in Liver Fibrosis. Front. Biosci. 2002, 7, d808–d826.
- Gandhi, C.R. Hepatic Stellate Cell Activation and Pro-Fibrogenic Signals. J. Hepatol. 2017, 67, 1104–1105.
- Schwabe, R.F.; Bataller, R.; Brenner, D.A. Human Hepatic Stellate Cells Express CCR5 and RANTES to Induce Proliferation and Migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G949–G958.
- Friedman, S.L. Molecular Regulation of Hepatic Fibrosis, an Integrated Cellular Response to Tissue Injury*. J. Biol. Chem. 2000, 275, 2247–2250.
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental Models of Liver Fibrosis. Arch. Toxicol. 2016, 90, 1025–1048.
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411.
- Xu, L. Human Hepatic Stellate Cell Lines, LX-1 and LX-2: New Tools for Analysis of Hepatic Fibrosis. Gut 2005, 54, 142–151.
- Taimr, P.; Higuchi, H.; Kocova, E.; Rippe, R.A.; Friedman, S.; Gores, G.J. Activated Stellate Cells Express the TRAIL Receptor-2/Death Receptor-5 and Undergo TRAIL-Mediated Apoptosis. Hepatology 2003, 37, 87–95.
- De Oliveira da Silva, B.; Alberici, L.C.; Ramos, L.F.; Silva, C.M.; da Silveira, M.B.; Dechant, C.R.P.; Friedman, S.L.; Sakane, K.K.; Gonçalves, L.R.; Moraes, K.C.M. Altered Global MicroRNA Expression in Hepatic Stellate Cells LX-2 by Angiotensin-(1–7) and MiRNA-1914–5p Identification as Regulator of pro-Fibrogenic Elements and Lipid Metabolism. Int. J. Biochem. Cell Biol. 2018, 98, 137–155.
- Zhao, X.-Y.; Zeng, X.; Li, X.-M.; Wang, T.-L.; Wang, B.-E. Pirfenidone Inhibits Carbon Tetrachloride- and Albumin Complex-Induced Liver Fibrosis in Rodents by Preventing Activation of Hepatic Stellate Cells. Clin. Exp. Pharmacol. Physiol. 2009, 36, 963–968.
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-Smooth Muscle Actin Expression Upregulates Fibroblast Contractile Activity. MBoC 2001, 12, 2730–2741.
- Tanaka, Y.; Nouchi, T.; Yamane, M.; Irie, T.; Miyakawa, H.; Sato, C.; Marumo, F. Phenotypic Modulation in Lipocytes in Experimental Liver Fibrosis. J. Pathol. 1991, 164, 273–278.
- Hautekeete, M.L.; Geerts, A. The Hepatic Stellate (Ito) Cell: Its Role in Human Liver Disease. Virchows Archiv 1997, 430, 195–207.
- Sato, M.; Suzuki, S.; Senoo, H. Hepatic Stellate Cells: Unique Characteristics in Cell Biology and Phenotype. Cell Struct. Funct. 2003, 28, 105–112.
- Stefanovic, B.; Hellerbrand, C.; Holcik, M.; Briendl, M.; Liebhaber, S.A.; Brenner, D.A. Posttranscriptional Regulation of Collagen alpha1(I) MRNA in Hepatic Stellate Cells. Mol. Cell. Biol. 1997, 17, 9.
- Varela-Rey, M.; Montiel-Duarte, C.; Osés-Prieto, J.A.; López-Zabalza, M.J.; Jaffrèzou, J.P.; Rojkind, M.; Iraburu, M.J. P38 MAPK Mediates the Regulation of A1(I) Procollagen MRNA Levels by TNF-α and TGF-β in a Cell Line of Rat Hepatic Stellate Cells11The Opinions or Assertions Contained Herein Are the Private Views of the Authors and Are Not to Be Construed as Official or as Reflecting the Views of the Department of the Army or the Department of Defense of the US. FEBS Lett. 2002, 528, 133–138.
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic Stellate Cell Lipid Droplets: A Specialized Lipid Droplet for Retinoid Storage. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2009, 1791, 467–473.
- Testerink, N.; Ajat, M.; Houweling, M.; Brouwers, J.F.; Pully, V.V.; van Manen, H.-J.; Otto, C.; Helms, J.B.; Vaandrager, A.B. Replacement of Retinyl Esters by Polyunsaturated Triacylglycerol Species in Lipid Droplets of Hepatic Stellate Cells during Activation. PLoS ONE 2012, 7, e34945.
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612.
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504.
- Dunning, S.; ur Rehman, A.; Tiebosch, M.H.; Hannivoort, R.A.; Haijer, F.W.; Woudenberg, J.; van den Heuvel, F.A.J.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Glutathione and Antioxidant Enzymes Serve Complementary Roles in Protecting Activated Hepatic Stellate Cells against Hydrogen Peroxide-Induced Cell Death. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 2027–2034.
- Byron, A.; Humphries, J.D.; Humphries, M.J. Defining the Extracellular Matrix Using Proteomics. Int. J. Exp. Pathol. 2013, 94, 75–92.
More