Lymphoproliferative disorders (LPD) are conditions characterized by the uncontrolled proliferation of B or T-cell lines. They encompass a wide spectrum of abnormalities, which may be broadly classified as reactive processes or malignant diseases, such as lymphoma, based on their cellular clonality and clinical behavior. While some of these disorders are rare, they may be encountered sporadically in clinical practice, causing diagnostic dilemmas owing to overlap in their clinical and imaging features with more common disorders.
21.1. Primary Central Nervous System Lymphoma with Attention to Lymphomatosis Cerebri: An Extremely Rare Diffuse B-Cell Lymphoma Variant
Primary central nervous system lymphoma (PCNSL), including the extreme rare variant lymphomatosis cerebri (LC), is a rare subtype of NHL with exclusive involvement of the central nervous system (CNS), representing 1% of all intracranial malignancies
[1][7]. LC implies the involvement of at least three cerebral lobes or three anatomical locations of the CNS, infiltrating the cerebral cortex and white matter tracts by lymphomatous cells, and some feel that LC would be more accurately named as a diffuse PCNSL variant
[2][8]. Historically, PCNSL has been associated with immunodeficiency
[3][9]. However, the incidence of PCNSL is increasing in immunocompetent patients, particularly among immunocompromised patients over 60 years of age
[4][10]. There is an association of PCNSL with Epstein–Barr virus (EBV) infection that may account for at least a portion of PCNSL cases in immunocompetent patients
[5][11]. In the previous 2008 WHO classification, it was hypothesized that the increasing incidence of PCNSL was falsely increased due to increased surveillance artifacts; however, in the current 4th edition of the WHO, an increasing incidence of PCNSL over the past two decades has been confirmed
[6][5].
The majority of PCNSLs including LC are diffuse large B-cell lymphomas (DLBCLs), with the remaining cases representing low-grade BCL (8%), Burkitt (5%), or TCL (2–3%)
[2][7][8,12]. Microscopically, LC is seen as a highly proliferative compact population of blastic B-lymphocytic cells with large pleomorphic nuclei and distinct nucleoli with predominant infiltration of the white matter and neuronal tracts without the formation of definite tumor or masses as seen in classic PCNSL. These lymphocytes showed the typical angiocentric infiltration pattern, and tumor cells invaded the neural parenchyma with a diffuse growth pattern forming perivascular cuffs
[7][12]. Unlike in PCNSL, cerebrospinal fluid (CSF) analysis from lumbar puncture is usually non-specific as LC rarely involves the meninges. LC can be classified into primary and secondary types based on differences in CNS infiltration at initial presentation. Primary LC displays CNS infiltration at presentation, whereas secondary LC shows progressive infiltration over time but no infiltration at presentation
[2][7][8,12].
Clinically, in both PCNSL and LC, the median age of presentation is 65 years, and in PCNSL most patients present clinically with focal neurological deficits. However, the most common presenting symptoms of LC are the impairment of cognitive functions with rapidly progressing dementia, behavioral abnormalities, and, less commonly, focal neurological deficits. The diagnosis of LC is usually delayed as clinical manifestations of LC are vague and overlap with imaging features of other disorders. The prognosis of LC is usually poor given the rapidly progressive disease process, and most LC patients do not survive beyond six months of initial presentation
[7][12].
The classic imaging appearance of PCNSL is a focal solitary mass or multiple masses within the brain parenchyma. On CT, PCNSL often shows homogenously hyperdense lesions due to high tumor cellularity with homogenous enhancement and a predilection for the deep gray matter and deep nuclei, periventricular trigon, and corpus callosum that often abuts the sub ependymal/ventricular or meningeal surfaces
[3][9]. Involvement of the meninges is most commonly seen in secondary lymphoma but can also be seen in PCNSL
[8][13]. On MRI, PCNSL tumors tend to be hypointense to isointense on T1-weighted sequences (T1W) and isointense to hyperintense on T2-weighted sequences (T2W) with intense enhancement on post-contrast images (
Figure 1).
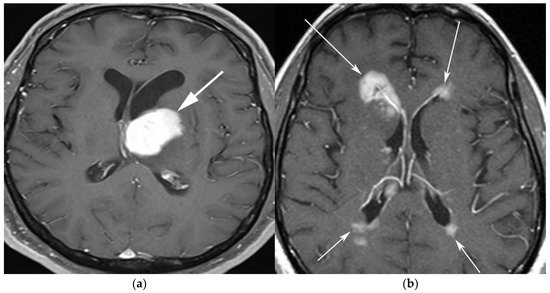
Figure 1. A 65-year-old man presenting with headache. (a) Axial T1 post-contrast fat-suppressed image shows homogenous enhancing mass within the left thalamus (white arrow) with mass effect on the 3rd ventricle and left putamen laterally. (b) Axial T1 post-contrast fat-suppressed images in a different patient show infiltrative periventricular soft tissue masses (white arrows). Both cases were pathologically proven primary central nervous system lymphoma-diffuse large B-cell histologic subtype.
Special sequences such as apparent diffusion coefficient (ADC) maps help differentiate PCNSL from high-grade tumors such as glioblastoma multiforme (GBM). PCNSL demonstrates marked diffusion restriction on diffusion-weighted images (DWI) with lower ADC values compared to GBM due to tumor restriction in lymphoma cells. The main role of 18F-FDG PET/CT in CNS lymphoma is to differentiate PCNSL vs. disseminated systemic lymphoma involving the CNS
[9][14]. Additionally, 18F-FDG PET/CT can help in distinguishing PCNSL from mimicking disorders such as hypometabolic intracranial opportunistic infections including CNS toxoplasmosis
[10][11][12][13][15,16,17,18]. Imaging findings of LC include diffuse, non-mass-like lesions within both hemispheres involving the white matter, corticospinal tracts (CST), and deep gray matter. These lesions show patchy contrast enhancement and diffuse abnormal T2W hyperintensity. Unlike PCNSL, LC shows variable restriction on DWI
[14][19]. Regions commonly affected in LC include the subcortical, deep, and periventricular white matter, CST, U-fibers, corpus callosum, and gray matter, with less frequent involvement of the spinal cord compared to PCNSL
[7][12]. Contiguous spread of lesions from the cerebral white matter to the brain stem along the corticospinal tract (CST) is a notable feature of LC, which can involve both the brain and brain stem (
Figure 2 and
Figure 3). Unlike PCNSL, the characteristic neuroradiological findings of LC with 18F-FDG PET/CT have not yet been established; nevertheless, they are usually seen as hypermetabolic mass like lesions
[2][15][8,20]. MRI spectroscopy (MRS) may help differentiate LC from other neurodegenerative or inflammatory conditions. On MRS, evidence of high cell membrane turnover (high choline peak), neuronal damage (decreased NAA levels), high lactate levels, and elevated lipids is typically demonstrated. Although many of these MRS features resemble high-grade gliomas and metastases, elevated lipids have been shown to be potentially useful to differentiate between these entities and LC
[16][21]. The diagnosis of lymphomatosis cerebri should be considered in patients presenting with a rapidly progressive decline in cognitive function, dementia, or behavioral abnormalities. Imaging findings including diffuse bilateral hemispheric cortical spinal tract involvement and pathology may show predominant infiltration of the neuronal tracts without the formation of definite masses. It is crucial that diagnosis of LC is only made after the exclusion of other common etiologies as the clinical and imaging findings of LC can be misattributed to other diffuse infiltrative brain tumors, leukoencephalopathy, vasculopathy, degenerative disease, ischemic processes, infectious processes, and toxic demyelinating diseases in addition to dementias and other psychiatric disorders such as depression
[2][7][8,12].
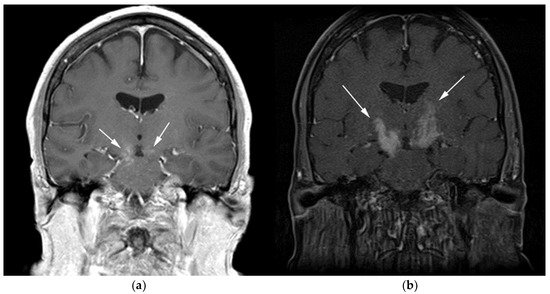
Figure 2. A 64-year-old-woman presenting with rapid onset of memory deterioration and altered sensorium. (a) Coronal MRI T1 post-contrast images show subtle enhancement along bilateral deep nuclei. At this time, no diagnosis was determined. After 3 weeks, patient presented to emergency department with worsening symptoms. A follow-up MRI was performed. (b) Coronal MRI post-contrast fat-saturated images revealed progressive increase in extent of diffuse infiltrative enhancing masses (white arrows).
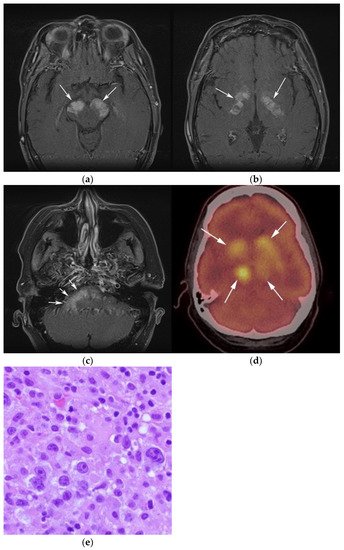
Figure 3. Same MRI series. (a) Axial post-contrast MRI image shows lesions extending caudally to involve the bilateral superior cerebral peduncles and anterior aspect of the mid-brain. (b) Axial MRI T1 post-contrast fat-saturated images show involvement of the posterior limb of the internal capsule and the thalami bilaterally (white arrows). (c) Extension along the lateral aspect of the pons, dentate nucleus, and the middle cerebellar peduncles (white arrows). (d) Axial FDG fused PET/CT image shows increased activity in the aforementioned lesions. Additional hypermetabolic lesions are seen along the course of the corticospinal tract (white arrows). Imaging findings and pattern of involvement are consistent with lymphomatosis cerebri. (e) H&E section shows a tumor composed of large and pleomorphic cells with intermingled small lymphocytes. Primacy CNS lymphoma typically demonstrates an angio-centric predilection. (Original magnification 400×, H&E stain). Other differential diagnoses include a vasculitic process, toxic or metabolic encephalopathy, paraneoplastic syndrome, or acute disseminated encephalomyelitis. Biopsy showed a large non-cohesive B-cell lymphocyte population consistent with LC.
2.2. Autoimmune Lymphoproliferative Syndrome
Autoimmune lymphoproliferative syndrome (ALPS) is a lymphoproliferative disorder that may be interpreted as malignant lymphoma. ALPS results from inherited mutations in the apoptosis signaling pathways
[17][18][22,23]. ALPS was first recognized in the early 1990s. Due to the overlap of clinical, pathologic, and imaging features with other malignant lymphoproliferative disorders, in addition to histopathology, a clinical diagnostic criterion for ALPS has been developed to help establish the diagnosis
[18][19][23,24]. The diagnostic criterion for ALPS includes both required and accessory criteria. Required criteria include chronic nonmalignant, noninfectious lymphadenopathy and/or splenomegaly (>6 months) and elevated CD3 + TCRab in the setting of normal or elevated lymphocyte counts with a lack of CD4 or CD8 expression, which are referred to as double-negative T cells (DNTs). Accessory criteria include somatic or germ-line pathogenic mutation (FAS, FASLG), elevated biomarkers (e.g., vitamin B-12), elevated IgG levels (polyclonal hypergammaglobulinemia), and family history of a nonmalignant/noninfectious lymphoproliferation with or without autoimmunity
[18][19][23,24]. A probable ALPS diagnosis can be entertained by the presence of the required criteria and any one of the secondary accessory criteria. Patients with probable ALPS undergo genetic or apoptosis assay to confirm the diagnosis
[19][24].
Clinically, ALPS presents in childhood and results in massive hepatosplenomegaly, chronic lymphadenopathy, thymic enlargement, and, in most patients, autoimmune phenomena such as hemolytic anemia
[20][25].
Imaging is not necessary to establish a diagnosis of ALPS but might aid in its diagnostic evaluation. Stability in the size of lymphadenopathy and hepatosplenomegaly over many years may suggest ALPS from other malignant lymphoproliferative diseases
[21][26] (
Figure 4).
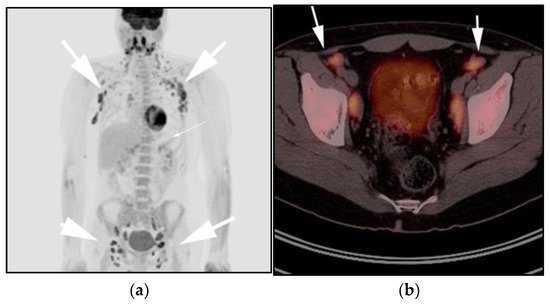
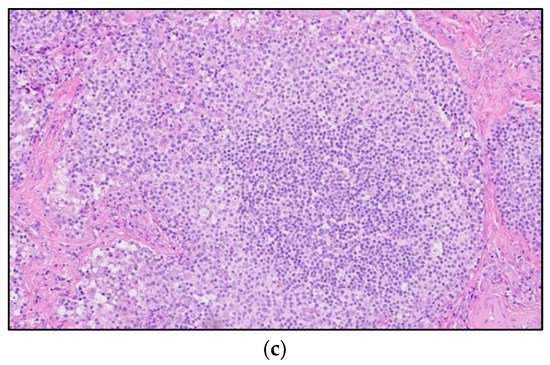
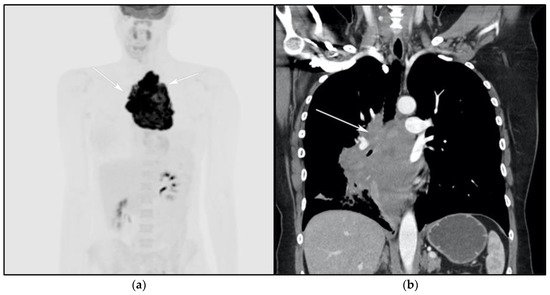
Figure 4. A 30-year-old male with known history of autoimmune lymphoproliferative disorder since childhood. (a) Maximal intensity projection (MIP) FDG PET/CT image shows generalized hypermetabolic lymphadenopathy throughout the head, neck, chest, abdomen, and pelvis (white arrows). Note that splenic activity is not visualized (thin white arrow) because the spleen was surgically removed during childhood due to splenomegaly. (b) Axial FDG PET image shows hypermetabolic adenopathy within pelvis mainly about the bilateral pelvic side wall and external iliac nodes (white arrows). Lymph nodes often show little or only modest uptake, a clue, in addition to stability of nodal size and over multiple time points. Biopsy is the only method for definitive diagnosis. (c) H&E section of a lymph node shows highly proliferative paracortical expansion composed of small proliferative lymphocytes and immunoblasts with preserved lymphoid follicular architecture. There is sinus histiocytosis. (Original magnification 400×, H&E stain).
Once the diagnosis of ALPS is established, baseline and periodic follow-up CT scans should therefore be obtained to document the stability of lymphadenopathy and hepatosplenomegaly. 18F-FDG PET/CT may also be helpful because mild to moderate FDG uptake is typical of ALPS compared to the high FDG uptake characteristic of malignant lymphomas. By evaluating for areas of high FDG uptake, 18F-FDG PET/CT may help in the early detection of lymphoma transformation and demonstrate the best site for biopsy, when indicated
[18][23]. Awareness of ALPS is pivotal because ALPS often requires long-term immunosuppressive therapies as opposed to chemotherapy that is standard for malignant lymphoproliferative disorders.
2.3. Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMLBCL) is an uncommon B-cell lymphoma, previously classified as a subtype of DLBCL but now considered an aggressive neoplasm that originates from thymic B cells, constituting less than 3% of all NHL cases
[22][27]. Classic HL (CHL) is always part of the clinical and imaging differential diagnosis of PMLBCL given the histologic overlap and many shared clinical features.
The median age of presentation with PMLBCL is slightly older than CHL, commonly affecting middle-aged women in their third or fourth decade. PMLBCL has a more favorable survival than other subtypes of aggressive DLBCLs, and poorer prognosis compared to CHL. The 5-year survival rate seen in cases of PMLBCL is 85%
[23][28]. Patients usually presents with local mediastinal compressive symptoms such as cough, chest pain, dyspnea, and vascular compressive symptoms such as superior vena cava syndrome. Factors that predict poor performance include male sex, old age, extension into the adjacent surrounding thoracic viscera such as pleural and pericardium, the presence of extra mediastinal extranodal disease (kidneys, adrenal glands, liver) at diagnosis, and/or early inadequate response to induction therapy
[22][23][27,28].
The genetic profile of PMLBCL is more similar to CHL than other BCLs
[24][3]. It is crucial for pathologists to be able to differentiate PMLBCL histologically. Microscopically, CHL histologic features usually show abundant infiltration with large, Hodgkin, and Reed–Sternberg cells (HRS) in a polymorphous background composed of small lymphocytes, macrophages, eosinophils, and plasma cells. HRS cells typically show expression of CD30 and CD15 and lack expression of CD20 and CD45. In PMLBCL, samples usually show diffuse proliferation of medium to large B cells associated with sclerosis and a degree of compartmentalization in more than 50% of cases. Lymphocytes usually express CD45 and CD3. CD15 is often absent and more heterogenous compared to HL. CD10 is often negative. Another differentiating feature is MAL protein positivity, which is a glycolipid-enriched membrane involved in the tumorigenesis of PMLBCL
[22][27]. Cases that cannot be differentiated from CHL after an extensive immunohistochemical workup should be categorized in the newly designated category: B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL (previously designated as “Gray-zone lymphomas”)
[23][28].
On imaging, patients typically present with a large ill-defined anterior mediastinal mass that often measures more than 10 cm in diameter with local infiltration of the underlying mediastinal structures
[25][26][27][29,30,31]. On CT, PMLBCL displays mixed attenuation due to the presence of necrosis and hemorrhage with heterogeneous post-contrast enhancement. On MRI, PMLBCL masses may show low signal on T1WI and variable signal on T2WI. After receiving chemotherapy, tumors tend to show decreased size with low signal on both T1WI and T2WI due to a fibrotic response.
The differential diagnosis of PMLBCL includes other anterior mediastinal masses such as CHL, germ cell tumors, and large thymic tumors, which can display necrosis and calcifications on CT
[28][32]. Differentiation between PMBCL and CHL is not possible based on imaging findings alone. More aggressive clinical behavior such as superior vena cava syndrome may help distinguish PMBCL from CHL
[29][33] (
Figure 5).
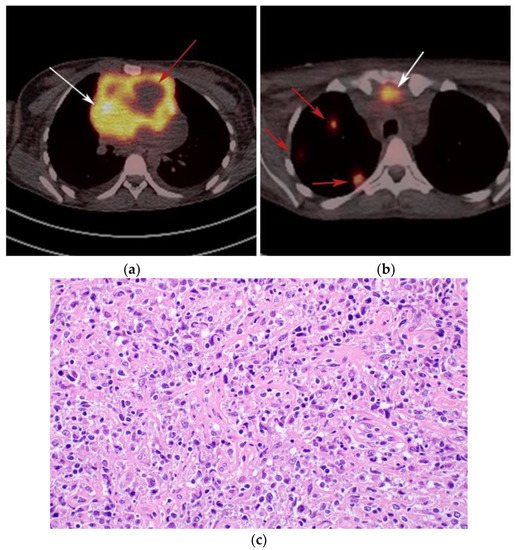
Figure 5. A 24-year-old female presenting with cough and dyspnea with plethora of the face. (a) Maximal intensity projection (MIP) FDG PET/CT shows hypermetabolic anterior mediastinal mass (white arrows) with no other sites of disease involvement in the body. (b) Coronal contrast-enhanced CT image shows significant vascular and airway compromise by an ill-defined anterior mediastinal mass (white arrow). Biopsy-proven primary mediastinal B-cell lymphoma. Main differential diagnosis would be Hodgkin’s lymphoma. However, unlike in Hodgkin’s lymphoma, primary mediastinal B-cell lymphoma often shows frequent invasion of the mediastinal vessels, frequently resulting in superior vena cava syndrome.
The use of 18F-FDG PET/CT is essential in the evaluation of patients with PMLBCL to reveal sites of disease not visible on CT and to provide more accurate staging and radiation field planning (
Figure 6). 18F-FDG PET/CT may also be beneficial for restaging after chemotherapy and/or radiotherapy, or when relapse is suspected
[30][34]. Negative 18F-FDG PET/CT after two or four cycles of chemotherapy has a negative predictive value and may predict excellent outcome in patients, achieving complete response without relapse. Patients who have residual activity equal to or higher than liver activity after immunochemotherapy treatment are more likely to relapse. In such instances, the addition of radiotherapy to the treatment regimen should be considered to avoid relapse in those high-risk patients. Relapse usually occurs within 1 year and is more likely to be widespread, involving distant extranodal sites such as the CNS, liver, kidneys, adrenal glands, GI tract, ovaries, and pancreas. Late relapses are very uncommon
[31][35]. 18F-FDG PET/CT can also efficiently assess post-treatment response, differentiating between necrotic or fibrotic tissue and residual masses containing viable tumor
[29][32][33,36]. There are various potential challenges to 18F-FDG PET/CT post-treatment implementation, including false-positive results secondary to thymic rebound hyperplasia, specifically seen in the young population. This can be limited by increasing the interval between treatment and imaging. Additionally, MRI can be helpful in those cases, and high signal on T1 in phase imaging with loss of signal on the out of phase sequences is consistent with thymic rebound hyperplasia (
Figure 7 and
Figure 8)
[30][33][34,37].
Figure 6. Imaging in a different patient with known diagnosis of primary mediastinal B-cell lymphoma. (a) Axial FDG PET/CT images show the hypermetabolic anterior mediastinal mass (white arrows), and there is also central areas of absent metabolic activity within the mass (red arrow), correlating with areas of fibrosis, a finding that is essential in prebiopsy planning to avoid false negative results. (b) Axial FDG PET/CT images show hypermetabolic right upper paratracheal nodes (white arrows) and several additional hypermetabolic pulmonary and pleural-based nodules within right upper lung (red arrows). (c) H&E section shows that the tumor is composed of large, atypical cells with reniform or multi-lobulated nuclei with abundant clear cytoplasm. Note the lymphoma cells are compartmentalized by the prominent sclerotic bands of fibrosis. (Original magnification 400x, H&E stain).
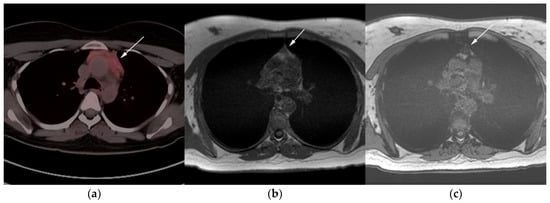
Figure 7. Imaging in a 20-year-old female with history of classic Hodgkin’s lymphoma (CHL) who had 4 cycles of chemotherapy. (a) Axial 18F-FDG PET/CT showing residual wedge-shaped/triangular activity seen within the anterior mediastinum (arrow). There was concern for residual disease vs. thymic rebound hyperplasia. (b) MRI axial T1 in phase and (c) MRI axial T1 out of phase show intermedial signal on T1 in phase (arrow) with dropped signal on the out of phase sequence (arrow), consistent with fat content due to thymic rebound hyperplasia.
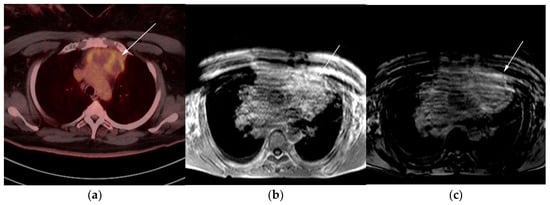
Figure 8. Imaging in a 60-year-old female presenting with ill-defined anterior mediastinal mass. (a) Axial 18F-FDG PET/CT showing ill-defined hypermetabolic mass within the anterior mediastinum (arrow). This was biopsy-proven thymic carcinoma. (b) MRI T1 in phase sequence, (c) MRI T1 out of phase—note there is no dropped signal on the T1 out of phase (arrows) due to lack of fat. This is consistent with residual disease.