Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Selene Pérez García and Version 2 by Jessie Wu.
Fibronectin is a component of the extracellular matrix essential to its assembly, which also regulates some cellular functions. However, cleavage of fibronectin in pathological conditions releases fibronectin fragments with pro-inflammatory and degradative properties. During the development of osteoarthritis, tissue proteolysis and injury induce extracellular matrix degradation, generating fibronectin fragments that promote inflammation and degradation by the induction of cytokine and proteinase expressions.
- Osteoarthritis
- Fibronectin
1. Introduction
Osteoarthritis (OA) is the most prevalent arthritic disease affecting the joints. While mainly related to aging, it is also associated with a diversity of risk factors including genetic predisposition, epigenetic factors, gender, obesity, exercise, work-related injury, and trauma. Irreversible and gradual loss of the articular cartilage remains the fundamental feature of OA pathophysiology [1][2][3][4][1,2,3,4]. However, in the course of the disease, all the joint tissues including synovium and bone undergo physical, functional, and metabolic alterations that comprise different cellular types as well as components of the extracellular cellular matrix (ECM). The ECM is a complex and specialized three-dimensional macromolecular network, present in nearly all tissues, which also interacts with cell surface receptors on joint resident cells. It is secreted, assembled, and modeled by the surrounding cells, providing physical support and organization to tissues. ECM is involved in many cell functions, providing cells with chemical and mechanical signals to regulate cell proliferation, survival, migration, and differentiation. However, changes in the composition and physical properties of the ECM lead to the development of many diseases, including cancer and rheumatic diseases, such as OA and rheumatoid arthritis (RA), among others [5][6][5,6]. Moreover, cell stress and ECM degradation promote maladaptive healing reactions, including inflammatory pathways of innate and adaptive immunity [7]. The loss of the biomechanical properties of cartilage induced by prior injuries or increased loading is the most significant feature in the initiation and progression of OA. Among the fibrillary components of ECM, the glycoprotein fibronectin (Fn) is an important member that acts as a bridging molecule in matrix assembly and cell–matrix interfaces. During the development of OA, tissue proteolysis and injury induce ECM degradation generating Fn fragments (Fn-fs), among other catabolic mediators, that promote inflammation and degradation by the induction of cytokine [8] and proteinase expressions [5].
2. Fibronectin
2. Fibronectin
Fibronectin is an adhesive glycoprotein, widely distributed in most ECM that regulates different cellular functions such as adhesion, motility, growth, differentiation, and opsonization [9][10][9,10]. It is a dimeric protein formed by two polypeptide chains with multiple repeated modular structures joined by two anti-parallel S-S bonds at the C-terminus. Each monomer of the Fn dimer has a molecular weight of 230–270 kDa, and it is composed of three different types of modules: twelve Fn type I (FnI), two Fn type II (FnII) and fifteen to seventeen Fn type III (FnIII) [11][12][13][14][15][11,12,13,14,15] (Figure 1A). FnI, FnII, and FnIII domains are made up of 40, 60, and 90 amino acids, respectively. Twenty different Fn proteins are observed in humans even though Fn is encoded by a single 75 kb gene [16]. This protein diversity is obtained by alternative splicing of two type FnIII exons, called extra domains A and B (EIIIA and EIIIB) and by a segment connecting two other FnIII repeats, FnIII14 and FnIII15, called the type III connecting segment (IIICS) or V domain, that can be assembled in four different ways or fully omitted. The multimodular structure and intermodular regions permit flexibility of the Fn molecule, which is involved in regulating its functions [13][15][16][13,15,16]. The different specific domains of Fn can interact with multiple binding partners, for example cell-surface receptors or other ECM components such as heparin, collagen, and proteoglycans. Fn is structured into four functional domains including a N-terminal domain (FnI1-9 plus FnII1-2), which binds Fn to heparin, collagen or fibrin; a central binding domain (FnII1-12) is responsible in part for Fn binding with the cells through the interaction with different integrins; and a C-terminal binding domain (FnIII12-14 plus IIICS plus FnI10-12) that also has other cell-binding sites, heparin- or fibrin-binding sites and the disulfide bridges responsible for Fn dimerization. For all of these reasons, Fn is considered to be a key molecule in the control of cellular regulatory processes and an essential scaffolding protein to preserve and maintain tissue organization [15].
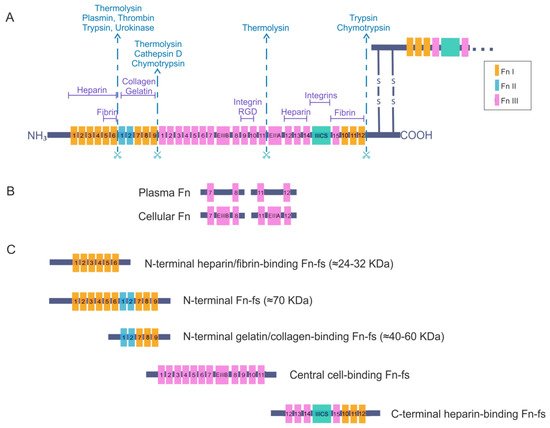
Figure 1. Fibronectin (Fn) and Fn fragments (Fn-fs) primary structure. (A) the schematic shows a representation of an Fn dimer and its interactions with different cell–surface receptors or other extracellular matrix (ECM) components such as heparin, collagen, and proteoglycans. Twelve Fn type I (FnI), two Fn type II (FnII) and fifteen constitutively expressed and two alternatively spliced Fn type III (FnIII) are indicated. The extra domains A and B (EIIIA and EIIIB) and the segment connecting two other FnIII repeats called the type III connecting segment (IIICS) or V domain are also shown. The scissors symbolize protease-sensitive regions of Fn and corresponding enzymes most commonly responsible for cleavage at these regions; (B) structural differences between plasma Fn and cellular Fn are shown; (C) the figure shows the different fragments of Fn and the structural domains that remain in each of them after breakage with proteolytic enzymes, adapted from [9][15][16][17][9,15,16,17].
There are two forms of Fn based on its solubility: plasma Fn, the soluble form, and cellular Fn, the water-insoluble form. The latter appears on cell surfaces and in the ECM of various tissues such as the synovial membrane and cartilage, but also in the synovial fluid. Moreover, plasma Fn has been reported to be integrated into the ECM with cellular Fn. Plasma Fn and cellular Fn have different structures and forms of assembly in three-dimensional networks (Figure 1B). Whereas plasma Fn lacks the alternatively spliced EIIIA and EIIIB regions, cellular Fn has different parts of these domains. In addition, only one subunit of plasma Fn possesses an IIICS segment. Taking these data into consideration, the number of different cellular Fn isoforms that can be generated is much higher than plasma Fn, due to the presence of alternative variants in the EIIIA, EIIIB, and IIICS segments [15][16][15,16].
Plasma Fn is synthesized by hepatocytes and directly released into circulation in a soluble, compact and inactive form. Blood plasma Fn levels increase after inflammation, major trauma, or pathologies such as atherosclerosis, ischaemic heart disease, and stroke [18][19][20][21][18,19,20,21]. Many cell types produce cellular Fn, including fibroblasts, endothelial cells, myocytes, chondrocytes, and synovial cells [15]. The different isoforms of cellular Fn are tissue-dependent, temporally regulated, and cell-type-specific. These isoforms regulate the properties of the ECM and consequently modulate different cellular processes. Under certain pathological conditions, some of the isoforms are exceptionally synthesized by cells or undergo a considerable increase in their synthesis. For instance, the isoform EIIIA is increased in synovial fluid of RA joints and correlates with the progression of joint destruction [22]. Another example is the presence of different synovial IIICS (+) isoforms, which vary their expression levels according to the degree of cartilage degeneration in OA [23]. An additional important fact to take into account is that the presence or absence of the different EIIIA, EIIIB, or IIISC regions affects the orientation and flexibility of the rest of the FnIII modules, altering the three-dimensional structure of the Fn and therefore its interactions during matrix assembly that can modulate Fn-cell signaling [15][24][15,24].
The functional form of Fn in vivo is its fibrillary state; thus, Fn molecules must be assembled into supermolecular fibers that form an interconnected network [14]. The presence of cells is necessary for Fn-matrix assembly, which occurs in several stages. First connections between Fn and cells are established through surface receptors such as integrins. Then, Fn is unfolded resulting in the exposure of Fn binding sites that allow Fn-Fn intermolecular interactions. Finally, the Fn-matrix assembly is formed. Both the expression of Fn and its assembly is regulated by a multitude of molecules in a cell-specific manner [15]. The Fn matrix assembly occurs at times of dynamic tissue remodeling, formation or repair and is essential during embryonic development [14].
3. Fibronectin and Fibronectin Fragments in Osteoarthritis
High levels of Fn have been described in the superficial layer of OA articular cartilage. Moreover, elevated levels of Fn accumulate in the inflamed synovial tissue and in the articular cartilage of RA joints. Enhanced levels of Fn are also observed in the synovial fluid, being 55% higher than levels in normal plasma [9][25][26][27][9,173,174,175]. In the course of these rheumatic diseases, in addition to the enhanced amounts of Fn in the joints, an exacerbated activation of Fn-degrading proteases has been described. In fact, increased levels of Fn-fs have been reported in the synovial fluid of OA and RA patients [9][17][28][29][9,17,176,177]. These fragments are included within the term matrikines, peptides originated from the fragmentation of ECM proteins that play an important role in both health and disease [30][178]. The activity of proteases is crucial during infection and inflammation, being responsible for the generation of different Fn-fs.
Fn-fs have properties not present in native Fn, and are the main candidates for the maintenance of cartilage destruction and synovial tissue inflammation in OA. Thus, 29-, 45-, 120-, and 200 kDa Fn-fs derived from Fn have been found in OA cartilage and synovial fluid, where they stimulate the production of various inflammatory cytokines, such as TNFα and IL-1β [31][114]. In addition, increased release of 29 kDa N-terminal heparin-binding, 50 kDa N-terminal gelatin-binding, and 110-140 kDa C-terminal heparin-binding Fn-fs have been detected in bovine injured cartilage explants compared to controls [32][179]. More recently, it has been reported that 29 kDa N-terminal heparin-binding Fn-fs inhibited autophagy through modifying localization of HMGB1 in human articular chondrocytes [33][180].
Along with OA, another musculoskeletal pathology with a huge worldwide prevalence is low back pain, which is generated by the degenerative disc disease, among other causes [34][181]. The degenerative progression in intervertebral discs is generally identified as intervertebral disc degeneration (IVD) [35][182]. IVD and OA share characteristics and properties such as cell physiology and ECM of the nucleus pulposus and articular cartilage. It has been suggested that, in acute stages, IVD is accompanied by loss of joint space, subchondral sclerosis, and osteophytes, comparable to OA in the articular joint. As occurs in OA, in IVD local inflammation is the result of mechanical overloading or low grade systemic inflammation, being characterized by increased levels of inflammatory cytokines. Moreover, this inflammatory cascade produces the degradation of ECM with the involvement of different proteinases including MMP-1–3, -7–10, and -12–14, as well as ADAMTS4 and -5. As a result, in both diseases, a chronic inflammation loop is established that degenerates both intervertebral disc and articular joint [36][183]. An additional element of similarity is the role that Fn plays in both pathologies. In this sense, an increased expression of Fn and Fn-fs has been reported in human spontaneously degenerative discs [37][184]. Moreover, the high levels of Fn-fs have been related with disc degeneration mediated by upregulation of MMP-3 and MMP-13 [38][29]. Ruel et al. have described that adult IVD tissues contains many Fn-fs that were absent from the infant disc tissue (6). The highest amount of FN-fs was present in the moderately degenerative discs, as well as in the initial phases of disc degeneration, before observable changes to the disc tissue arise, thus suggesting that Fn-fs play a significant role in the initiation and evolution of disc degeneration [39][185].
Baker et al. proposed a mathematical model showing that positive and negative feedback monitoring the degree of cytokine production can regulate either the pre-disposition to OA or the initiation of OA. Moreover, they proposed that manipulation of cytokines, proteinases and Fn-fs levels could be used to treat OA and other related pathologies, suggesting that multiple treatment targets may be essential to break or slow disease progression [40][186].
In the next section, we describe the multi-target effect of different Fn-fs in the modulation of ECM-degrading enzymes in OA.
3.1. Effect of Fibronectin Fragments in the Profile of OA Matrix-Remodeling Proteinases
During OA, ECM degrading enzymes induce the release of damage-associated molecular patterns (DAMPs) with catabolic properties, including Fn-fs. These Fn-fs act as pro-inflammatory mediators in a positive feedback loop inducing the expression of pro-inflammatory cytokines, nitric oxide (NO), and other inflammatory mediators, as well as proteinases, degrading ECM components, and therefore promoting inflammation and cartilage destruction in OA [9][28][29][9,176,177]. In this regard, different authors have described the induction of proteinases mediated by Fn-fs in chondrocytes and SF, including uPA, MMPs, and ADAMTSs [41][42][31][26][43][33,80,114,174,187] (Figure 14).
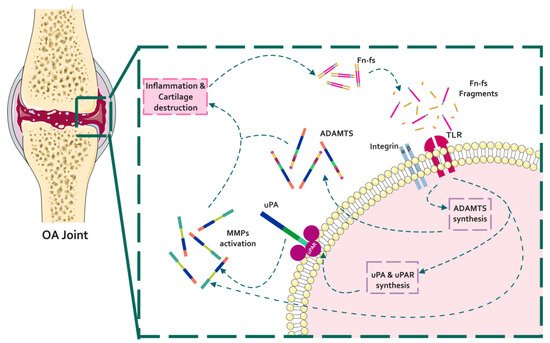
Figure 14. Schematic representation of the interaction of Fn with proteinases in the context of OA pathology. In a situation of inflammation and cartilage damage, Fn cleavage from the cartilage ECM promotes the release of Fn-fs to the joint microenvironment. Binding of Fn-fs to their receptors (integrins and TLRs) activates different signaling pathways increasing the expression of proteinases (uPA, MMPs, and ADAMTSs). In turn, active proteinases degrade the cartilage ECM inducing a feed-back loop of inflammation and cartilage degradation.
3.1.1. Fn-fs Modulate the uPA System and Induce MMP Expression
Deregulation of the uPA system is related to the development of several pathologies including OA [44][45][46][47][39,54,55,92]. During this disease, Fn-fs are released to the joint microenvironment as a consequence of ECM degradation. In this respect, it has been described that 45 kDa N-terminal gelatin-binding Fn-fs increase uPA expression and activity and uPAR production in OA-SF [42][80]. Furthermore, 29 kDa N-terminal heparin-binding Fn-fs also increases uPA levels in bovine articular cartilage [31][114]. Moreover, 40- to 60 kDa N-terminal gelatin-binding Fn-fs are associated with the binding of immunoglobulins and complement proteins to inflammation sites, as well as with the activity of tPA and uPA [17][42][17,80]. In addition, these fragments have been related to proteoglycanase activity, associated with MMPs or ADAMTSs [17][41][17,33].
As mentioned above, binding of active uPA to uPAR induces the activation of plasminogen to plasmin, which in turn activates MMPs. In this regard, in addition to the uPA system, 45 kDa N-terminal gelatin-binding Fn-fs also increase the production of MMP-9 and MMP-13 in OA-SF culture supernatants [42][80]. Moreover, these fragments induce MMP-3 and MMP-13 synthesis in porcine cartilage [48][188]. Elevated release of MMPs and proteoglycans, as well as increased proteoglycan degradation and induction of NO and catabolic cytokines, has been observed in bovine articular cartilage [31][114]. In addition, 50% removal of articular cartilage proteoglycans mediated by Fn-fs has been described in rabbit knee joints [49][189]. This proteoglycan depletion is mainly associated with the activity of MMP-3, increased by 29 kDa N-terminal heparin-binding, 50 kDa N-terminal gelatin-binding, and 140 kDa central cell-binding Fn-fs in bovine cartilage. MMP-3 in turn degrades Fn, generating new Fn-fs in a positive feedback loop [31][32][50][114,179,190]. Furthermore, 29 kDa N-terminal heparin-binding Fn-fs also enhance gelatinase expression in bovine cartilage [31][26][114,174]. These Fn-fs have been involved in the stimulation of the expression of catabolic factors through the TLR2-dependent signaling pathway. Thus, the exposure to 29 kDa N-terminal heparin-binding Fn-fs from synovial fluid of OA patients, increased MMP-1, MMP-3, and MMP-13 expression in primary chondrocytes, where TLR2 knockdown significantly blocked the synovial fluid-induced MMP stimulation [51][28]. Moreover, involvement of α5 integrins has been shown in cartilage proteoglycan degradation. In this respect, α5 integrins seem to be implicated in the N-terminal heparin- and gelatin-binding, central cell-binding, and C-terminal heparin-binding Fn-fs induction of proteoglycan loss, promoting cartilage destruction in bovine cartilage [52][26][53][25,174,191].
Central cell-binding Fn-fs are able to stimulate MMP-1 and MMP-3 expression in rabbit SF [43][187], as well as MMP-3 in rabbit chondrocytes [54][192]. Moreover, 120 kDa central cell-binding Fn-fs increase MMP-13 in human chondrocytes, with the involvement of α5β1 integrins [55][56][193,194]. These 120 kDa Fn-fs also induce collagenase expression in rabbit SF [57][195]. In addition, 29 kDa N-terminal heparin-binding and 140 kDa central cell-binding Fn-fs, increase the tissue inhibitor of metalloproteinases TIMP-1 in bovine articular cartilage [31][114].
Finally, C-terminal heparin-binding Fn-fs induce MMP-3 and MMP-13 production in bovine cartilage [58][196], MMP-1, MMP-2, MMP-9, and MMP-13 in human cartilage [59][197], as well as MMP-1, MMP-3, and MMP-13 in RA-SF [60][198]. Moreover, these Fn-fs also increase type II collagen cleavage in human and bovine cartilages, mainly mediated by MMP-13 [58][59][196,197].
While Fn is less active than its degradation products, it is also able to induce the expression of metalloproteinases in some contexts. In this regard, Fn increases MMP-9 production in human and murine macrophages [61][199]. Furthermore, Fn induces MMP-2 expression in human prostate cancer cells [62][200]. MMPs in turn cleave Fn, generating Fn-fs. Therefore, MMP-1, MMP-3, MMP-13, and MMP-14 degrade Fn generating 70 kDa Fn-fs, where MMP-13 and -14 were the most effective. Moreover, MMP-13 and -14, are also able to generate 52-, 40-, 32-, and 29 kDa Fn-fs in articular cartilage from bovine knee joints [63][201]. In addition, MMP-8, -9 and -12 also cleave Fn in human articular cartilage, where MMP-12 seems to be the main metalloproteinase [64][202].
3.1.2. Fn-fs Modifies ADAMTS Expression and Signaling
The 45 kDa N-terminal gelatin-binding Fn-fs has been shown to increase aggrecanase levels in SF. Specifically, these Fn-fs induce ADAMTS4 expression and protein production in HD- and OA-SF, ADAMTS5 expression and protein production in OA-SF, as well as aggrecanase activity in OA-SF, and GAGs release in HD- and OA-SF [41][33]. In addition, these 45 kDa Fn-fs induce aggrecan degradation and the generation of aggrecanase-derived neoepitopes in porcine cartilage [48][188]. Induction of ADAMTS5 mediated by 29 kDa N-terminal heparin-binding Fn-fs has also been described in human and bovine chondrocytes [65][66][203,204]. Furthermore, these ADAMTSs, primarily ADAMTS4, are also involved in Fn cleavage in human articular cartilage [64][67][202,205]. On the other hand, the 40 kDa C-terminal heparin-binding Fn-fs is able to inhibit the aggrecanase activity of ADAMTS4 in human chondrocytes, suggesting a positive effect of this Fn-fs by avoiding cartilage degradation [68][206].
Regarding COMP-degrading ADAMTSs, 45 kDa N-terminal gelatin-binding Fn-fs also increase ADAMTS7 expression and protein production in HD- and OA-SF, ADAMTS12 expression, and protein production in OA-SF, and COMP degradation in HD- and OA-SF [41][33]. On the other hand, concerning ECM assembly, COMP interacts with Fn, with a predominant binding site at the N-terminal domain of Fn [69][150].
In relation to other ADAMTSs, ADAMTS16 inhibits Fn fibrillogenesis and cleaves Fn, releasing a 30 kDa N-terminal heparin-binding Fn-fs, which in turn, upregulates MMP-3, which is also able to cleave Fn, generating new Fn-fs and inducing a positive degradative feedback loop in an epithelial cell line [70][207]. Additionally, ADAMTS9 is involved in Fn turnover and fibrillogenesis in ECM remodeling during mouse embryogenesis [71][208].
Finally, regarding signaling pathways implicated in ADAMTS expression, 45 kDa N-terminal gelatin-binding Fn-fs induce Runx2 activation in HD- and OA-SF and Wnt/β-catenin signaling in OA-SF [41][72][33,50]. Furthermore, Fn is a target of Wnt signaling in mouse embryonic lung morphogenesis [73][209] and, in turn, the canonical Wnt pathway is involved in Fn and metalloproteinase expression in RA-SF [74][210], suggesting a feedback loop between Fn and β-catenin [75][211].