The renin–angiotensin system (RAS), an essential enzymatic cascade involved in maintaining blood pressure and electrolyte balance, is involved in the pathogenicity of COVID-19, since the angiotensin-converting enzyme II (ACE2) acts as the cellular receptor for SARS-CoV-2 in many human tissues and organs. In fact, the viral entrance promotes a downregulation of ACE2 followed by RAS balance dysregulation and an overactivation of the angiotensin II (Ang II)–angiotensin II type I receptor (AT1R) axis, which is characterized by a strong vasoconstriction and the induction of the profibrotic, proapoptotic and proinflammatory signalizations in the lungs and other organs. This mechanism features a massive cytokine storm, hypercoagulation, an acute respiratory distress syndrome (ARDS) and subsequent multiple organ damage.
- SARS-CoV-2
- COVID-19
- ACE-2
- Ang II/AT1R axis
- RAS imbalance
- cytokine storm
- underlying diseases
- genetic polymorphisms
- host susceptibility factors
1. Introduction
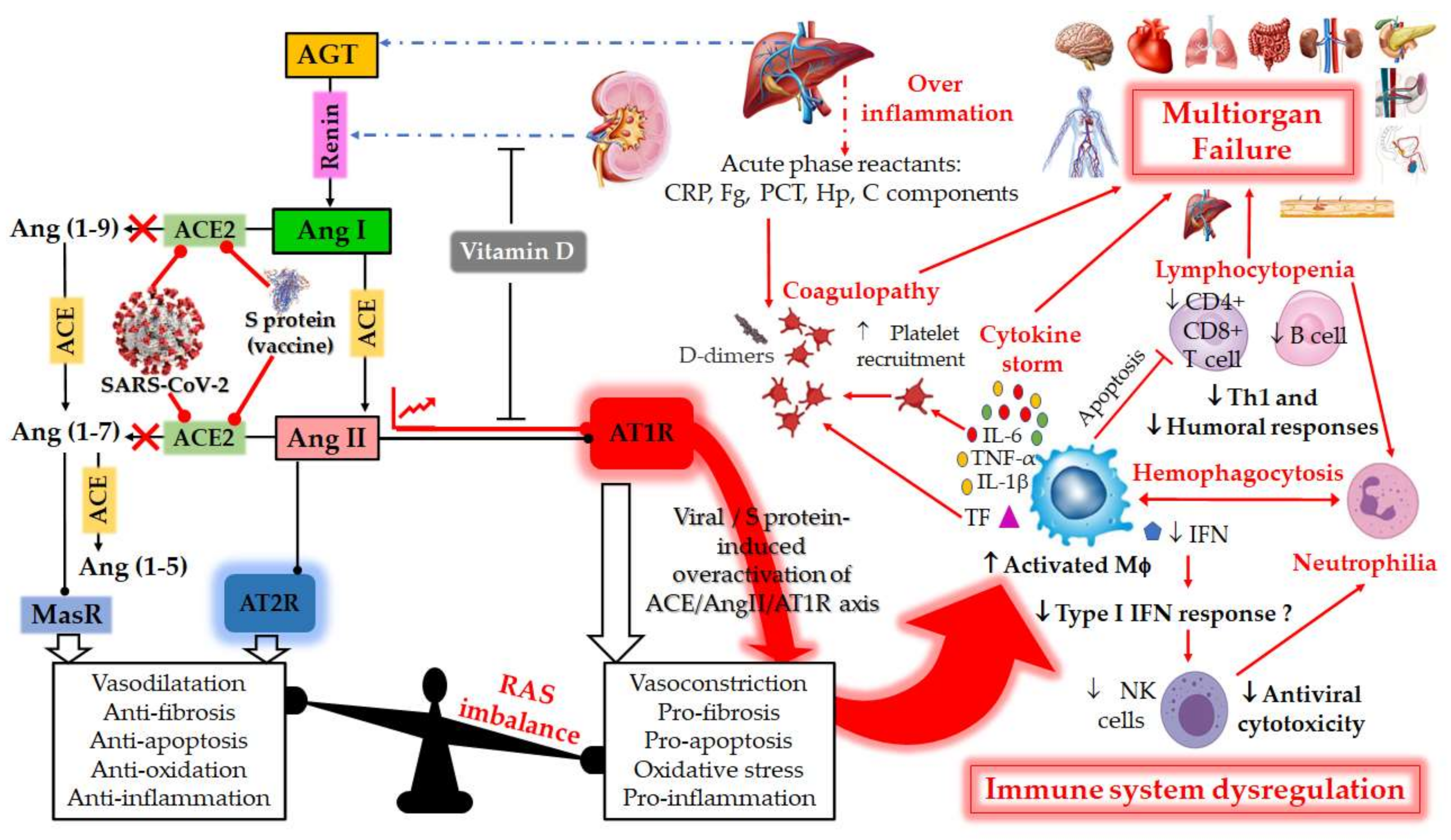 Figure 1. Schematic diagram of the dysregulation in the Renin–Angiotensin System (RAS) and the host immune system caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or by vaccination with mRNA encoding SARS-CoV-2 spike (S) glycoprotein. RAS is a metabolic cascade which supports a series of enzymatic reactions in which the liver secreted AGT is transformed into Ang I by renin, which is a protease secreted by juxtaglomerular kidney cells in response to decrease in blood pressure or sodium load in the distal convoluted tubule. Ang I is subsequently converted to Ang II by ACE which can bind to the AT1R to exert several actions, such as vasoconstriction, pro-fibrosis, pro-apoptosis, oxidative stress and pro-inflammation. ACE2 counterbalances Ang II/AT1R effects by cleaving Ang I and Ang II into Ang-(1–9) and Ang-(1–7), respectively. Ang-(1–9) is also converted into Ang-(1–7), a negative regulator of the RAS, which binds to the MAS receptor to exert protective actions of vasodilatation, anti-fibrosis, anti-apoptosis, anti-oxidative and anti-inflammation. Ang-II can also bind to AT2R to counteract the aforementioned effects mediated by AT1R. The balance between the Ang II/AT1R axis and the ACE2/Ang (1–7)/MasR axis is therefore maintained under physiological conditions. However, during SARS-CoV-2 infection or upon receiving a spike protein-based vaccine, the viral Spike (S) glycoprotein binding to ACE2 receptor induces overactivation of the ACE/Ang II/AT1R axis. This event prevents normal Ang II degradation, the excess of which leads to AT1R overactivation and RAS system imbalance. Such an imbalance is very deleterious for the human body, mainly due to the important immunomodulatory roles of ACE2, which can directly interact with macrophages in the setting of vascular and lung inflammation. Patients with severe COVID-19 infections show hallmarks of sepsis, widely explained by an exacerbation of macrophage activation, including excessive inflammation with the presence of acute phase reactants (such as D-dimer, CRP, etc.), impending cytokine storms and overexpression of IL-1β, IL-2, IL-6, and TNF-α in the early phase of the disease. These induce the production of a compelling number of factors linked to the coagulation cascade (TF, Fb, etc.) and resulting in the onset of thrombi and associated disseminated intravascular coagulation (DIC). The inflammatory response to SARS-CoV-2 also consists of lymphopenia occurring early in >80% of patients and is prognostic, manifested as reduction in—and functional exhaustion of—CD4+ more than CD8+ T cells. Such impaired T cell responses can result from deficient IFN production, as IFNs act on the antigen-presenting cells, T cells, and induce other cytokines and chemokines that regulate T-cell responses. These events lead to imbalance of the innate/acquired immune response, delayed viral clearance and unusual predominance of hyperstimulated macrophage and neutrophil in targeted injured tissues. The permanent immune activation in predisposed elderly adults and patients with cardiovascular risk can lead to hemophagocytosis-like syndrome, with uncontrolled amplification of cytokine production, leading to endothelial dysfunction, tissue damage and multiorgan failure, which is the starting point of a progression towards the serious and fatal complications of COVID-19. This syndrome results from the ineffective activation of cytotoxic CD8+ T lymphocytes and Natural Killer T lymphocytes, and leads to ineffective viral cytotoxicity and weak antibody production. NK cells are regulated by IFNs during coronavirus infection, and patients with severe COVID-19 showed profound depletion and functional exhaustion of NK cells, the dysfunction of which could be due to dysregulation of IFN responses. On the other side, Vitamin D could help avoid the potential deleterious COVID-19 effects sometimes observed following vaccination, by either inhibiting renin secretion or suppressing AT1R overactivation. AGT: angiotensinogen; Ang I: angiotensin I; ACE: angiotensin-converting enzyme; Ang II: angiotensin II; ACE-2: angiotensin-converting enzyme-2; Ang 1–7: angiotensin 1–7; AT1R: angiotensin II type 1 receptor; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; COVID-19: coronavirus disease 2019; CRP, c-reactive protein; IL-1β, interleukin-1β; IL-6, interleukin-6; TNF-α. Tumor Necrosis Factor-alpha; INF, Interferon; Fg, fibrinogen; PCT, procalcitonin; Hp, haptoglobin; C, complement; MΦ, macrophage; NK, Natural Killer; Th1, T helper type 1; TF, tissue factor.
Figure 1. Schematic diagram of the dysregulation in the Renin–Angiotensin System (RAS) and the host immune system caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or by vaccination with mRNA encoding SARS-CoV-2 spike (S) glycoprotein. RAS is a metabolic cascade which supports a series of enzymatic reactions in which the liver secreted AGT is transformed into Ang I by renin, which is a protease secreted by juxtaglomerular kidney cells in response to decrease in blood pressure or sodium load in the distal convoluted tubule. Ang I is subsequently converted to Ang II by ACE which can bind to the AT1R to exert several actions, such as vasoconstriction, pro-fibrosis, pro-apoptosis, oxidative stress and pro-inflammation. ACE2 counterbalances Ang II/AT1R effects by cleaving Ang I and Ang II into Ang-(1–9) and Ang-(1–7), respectively. Ang-(1–9) is also converted into Ang-(1–7), a negative regulator of the RAS, which binds to the MAS receptor to exert protective actions of vasodilatation, anti-fibrosis, anti-apoptosis, anti-oxidative and anti-inflammation. Ang-II can also bind to AT2R to counteract the aforementioned effects mediated by AT1R. The balance between the Ang II/AT1R axis and the ACE2/Ang (1–7)/MasR axis is therefore maintained under physiological conditions. However, during SARS-CoV-2 infection or upon receiving a spike protein-based vaccine, the viral Spike (S) glycoprotein binding to ACE2 receptor induces overactivation of the ACE/Ang II/AT1R axis. This event prevents normal Ang II degradation, the excess of which leads to AT1R overactivation and RAS system imbalance. Such an imbalance is very deleterious for the human body, mainly due to the important immunomodulatory roles of ACE2, which can directly interact with macrophages in the setting of vascular and lung inflammation. Patients with severe COVID-19 infections show hallmarks of sepsis, widely explained by an exacerbation of macrophage activation, including excessive inflammation with the presence of acute phase reactants (such as D-dimer, CRP, etc.), impending cytokine storms and overexpression of IL-1β, IL-2, IL-6, and TNF-α in the early phase of the disease. These induce the production of a compelling number of factors linked to the coagulation cascade (TF, Fb, etc.) and resulting in the onset of thrombi and associated disseminated intravascular coagulation (DIC). The inflammatory response to SARS-CoV-2 also consists of lymphopenia occurring early in >80% of patients and is prognostic, manifested as reduction in—and functional exhaustion of—CD4+ more than CD8+ T cells. Such impaired T cell responses can result from deficient IFN production, as IFNs act on the antigen-presenting cells, T cells, and induce other cytokines and chemokines that regulate T-cell responses. These events lead to imbalance of the innate/acquired immune response, delayed viral clearance and unusual predominance of hyperstimulated macrophage and neutrophil in targeted injured tissues. The permanent immune activation in predisposed elderly adults and patients with cardiovascular risk can lead to hemophagocytosis-like syndrome, with uncontrolled amplification of cytokine production, leading to endothelial dysfunction, tissue damage and multiorgan failure, which is the starting point of a progression towards the serious and fatal complications of COVID-19. This syndrome results from the ineffective activation of cytotoxic CD8+ T lymphocytes and Natural Killer T lymphocytes, and leads to ineffective viral cytotoxicity and weak antibody production. NK cells are regulated by IFNs during coronavirus infection, and patients with severe COVID-19 showed profound depletion and functional exhaustion of NK cells, the dysfunction of which could be due to dysregulation of IFN responses. On the other side, Vitamin D could help avoid the potential deleterious COVID-19 effects sometimes observed following vaccination, by either inhibiting renin secretion or suppressing AT1R overactivation. AGT: angiotensinogen; Ang I: angiotensin I; ACE: angiotensin-converting enzyme; Ang II: angiotensin II; ACE-2: angiotensin-converting enzyme-2; Ang 1–7: angiotensin 1–7; AT1R: angiotensin II type 1 receptor; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; COVID-19: coronavirus disease 2019; CRP, c-reactive protein; IL-1β, interleukin-1β; IL-6, interleukin-6; TNF-α. Tumor Necrosis Factor-alpha; INF, Interferon; Fg, fibrinogen; PCT, procalcitonin; Hp, haptoglobin; C, complement; MΦ, macrophage; NK, Natural Killer; Th1, T helper type 1; TF, tissue factor.2. RAS and COVID-19
2.1. RAS: A Portal Entry for SARS-CoV-2
2.2. RAS Imbalance and Underlying Pathologies
Since SARS-CoV-2 uses ACE2 as a key receptor for cellular entry, the latter subsequent downregulation will decrease Ang II degradation, leading therefore to its accumulation [17,33][17][33]. This contributes to a potential dysregulation and overactivation of RAS (Figure 1) [17,47][17][36]. Studies have shown that RAS imbalance worsens COVID-19 prognosis and aggravates its pathogenesis [17,46][17][37]. Therefore, the most common underlying pathophysiology of COVID-19 is a viral acute respiratory distress syndrome (ARDS) coupled with the cytokine storm syndrome [17]. According to current knowledge, the main cause of death in COVID-19 patients in intensive care unit (ICU) is the ARDS secondary to SARS-CoV-2 pneumonia [35]. ARDS is characterized by a hypoxemia with increased capillary–alveolar permeability, reduced lung compliance, alveolar epithelial cell loss, neutrophil infiltration and a diffuse bilateral pulmonary infiltrate that could lead to alveolar and interstitial remodeling and fibrosis [48][38]. This could lead to the loss of pulmonary perfusion regulation and hypoxic vasoconstriction, as well as a low ventilation perfusion ratio [2], thus requiring mechanical ventilation [48][38]. According to Richardson et al., 80% of patients who required mechanical ventilation after COVID-19 infection evolved to death, emphasizing that ARDS is an underlying pathophysiology in COVID-19 patients which may be responsible for the high mortality rates [49][39]. It has been shown that RAS imbalance may influence the pathogenesis of ARDS through Ang II and bradykinin [48][38]. Experiments have revealed that rats with knock-out ACE2 exposed to non-SARS lung damage (such as endotoxin) developed a severe ARDS compared to the wild type rats [50][40]. Therefore, ACE2 was shown to have a protective effect in rat models of acute lung injury, with ACE, Ang II and AT1R being considered as lung injury-promoting elements [50][40]. Imai et al., have shown an upregulation of Ang II by ACE in the pathogenesis of acute lung injury through the AT1a receptor, leading therefore to severe lung failure [28]. A study conducted by Kuba et al., showed that the blockage of ACE2 or its genetic manipulation leads to increased lung edema, vascular permeability and neutrophil accumulation [50][40].2.3. RAS Component Polymorphism
Many studies described the association of the RAS component genetic variation with the prevalence of COVID-19 diseases [47,68][36][41]. Therefore, it has been thought that genetic factors may render the host resistant or susceptible to infection with SARS-CoV-2 [47][36]. The prevalence and disease outcome were linked to ACE polymorphisms [36][42]. The genetic polymorphisms of ACE1 and ACE2 genes may change their levels of expression, therefore leading to an increase in capillary permeability, coagulation, fibrosis and apoptosis in alveolar cells [69][43]. In a pilot study conducted by Cafiero et al., it was found that some genetic variants in the RAS pathway may be potential actors for determining the clinical outcome and the pathological conditions associated COVID-19, such as DIC, interstitial pneumonia, thrombosis, conjunctivitis and the cytokine storm [35]. Thus, inflammation and lung injury caused by ACE2 decrease, following viral binding, could be negatively affected by ACE’s different genotypes, which in turn could increase ACE expression levels and then those of Ang II [36][42]. The knowledge of these polymorphisms could help the management of COVID-19 infected patients [36][42]. The major RAS component polymorphisms are illustrated in Table 1.| RAS Component Gene |
Chromosomal Location | Associated Disease/Phenotype |
Mutations, Polymorphisms and rs Number |
Allele/Genotype Frequencies in Populations and Ethnicities |
References | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACE1 | 17q23.3 | CVD Kidney disease Autoimmune diseases Hypertension Hypercoagulability ARDS Type 2 diabetes Risk of obesity |
Insertion/Deletion (I/D) of a 287-bp | Alu | repeat in intron 16 (rs1799752) |
Lebanese | I | : 0.27, | D | : 0.73 | [77] | [44] | ||||||||||||||||||||
| Indians | I | : 0.55, | D | : 0.45 | Whites | I | : 0.5, | D | : 0.5 | African Americans | I | : 0.41, | D | : 0.59 | [76] | [45] | ||||||||||||||||
| British | I | : 0.31, | D | : 0.69 (ARDS) | I | : 0.49, | D | : 0.51 (healthy population) | [73] | [46] |
||||||||||||||||||||||
| Chinese | I | : 0.705, | D | : 0.295 | [75] | [47] | ||||||||||||||||||||||||||
| Italians | I | : 0.342, | D | : 0.658 | [56] | [48] | ||||||||||||||||||||||||||
| Italians | I | : 0.27, | D | : 0.73 | [35] | |||||||||||||||||||||||||||
| Germans | I | / | I | : 0.27, | I | / | D | : 0.43, | D | / | D | : 30 | [48] | [38] | ||||||||||||||||||
| Indians | I | : 0.575, | D | : 0.425 | [78] | [49] | ||||||||||||||||||||||||||
| ACE2 | Xp22.2 | Cardiovascular risk, Retinopathy in type-2 Diabetes Mellitus, Hypertension and Hypertensive left ventricular hypertrophy | c.*1860-449C > T SNP (rs2074192) |
Italians | C | : 0.56, | T | : 0.44 | [35] | |||||||||||||||||||||||
| c.*264+788T > C (rs2106809) |
Italians | A | : 0.77, | G | : 0.33 | |||||||||||||||||||||||||||
| c.2115-268A > T SNP (rs233574) |
Africans | C | : 0.92, | T | : 0.08 | Europeans | C | : 0.67, | T | : 0.33 | East Asians | C | : 0.996, | T | : 0.004 | South Asians | C | : 0.814, | T | : 0.814 | Americans | C | : 0.767, | T | : 0.233 | [79] | [50] | |||||
| c.1402A > G p.Ile468Val SNP (rs191860450) |
East Asians | with an allele frequency (AF) = 0.011 | [16] | |||||||||||||||||||||||||||||
| c.1022A > G p.Lys341Arg SNP (rs138390800) |
Africans | AF = 4 × 10 | −3 | |||||||||||||||||||||||||||||
| c.2191C > T p.Leu731Phe SNP (rs147311723) |
Africans | AF = 0.014 |
||||||||||||||||||||||||||||||
| c.631G > A p.Gly211Arg SNP (rs148771870) |
Europeans | AF = 2 × 10 | −3 | South Asians | AF = 1.9 × 10 | −3 | ||||||||||||||||||||||||||
| c.2089A > G p.Arg697Gly SNP (rs751603885) |
South Asians | AF = 2.4 × 10 | −3 | |||||||||||||||||||||||||||||
| c.2074T > C p.Ser692Pro SNP (rs14903946) |
Africans | AF = 6 × 10 | −3 | |||||||||||||||||||||||||||||
| c.55T > C p.Ser19Pro SNP (rs73635825) |
Africans | AF = 3 × 10 | −3 | |||||||||||||||||||||||||||||
| AGT | 1q42.2 1q42–43 |
Hypertension Heart failure Myocardial infraction |
c.704T > C p.Met235Thr (aka Met268Thr) SNP (rs699) |
Tunisians | M | / | M | : 0.291, | M | / | T | : 0.291 | T | / | T | :0.419 | [80] | [51] | ||||||||||||||
| Vietnamese | T | : 0.92, | M | : 0.08 | [81] | [52] | ||||||||||||||||||||||||||
| Iranians | T | : 0.39, | M | : 0.61 | [82] | [53] | ||||||||||||||||||||||||||
| Indians | M | : 0.52, | D | : 0.48 | [78] | [49] | ||||||||||||||||||||||||||
| New Zealanders | T | / | T | : 0.19, | T | / | M | : 0.47, | M | / | M | : 0.34 | [83] | [54] | ||||||||||||||||||
| c.521C > T p.Thr174Met SNP (rs4762) |
New Zealanders | T | / | T | : 0.7, | T | / | M | : 0.2, | M | / | M | : 0.1 | [83] | [54] | |||||||||||||||||
| AT1R | 3q21–q25 | Systolic blood pressure Left ventricular hypertrophy Hypertension Aortic stiffness Myocardial infarction Carotid intimal-medial thickening, CAD and stroke, Overweight, Diabetes |
c.1166A > C in the 3′ UTR SNP (rs5186) |
Egyptians | C | :0.24, | A | :0.76 (control group) | C | :0.34, | G | :0.66 (premature CAD patients) | [84] | [55] | ||||||||||||||||||
| Jordanians | Higher frequency of | A | allele | [85] | [56] | |||||||||||||||||||||||||||
| Iranians | Higher frequency of | A | allele | [86] | [57] | |||||||||||||||||||||||||||
| AT2R | Xq23–26 | Metabolic syndrome | −1332A > G SNP (rs1403543) |
Egyptians | A | :0.55, | G | :0.45 (control group) | A | :0.41, | G | :0.50 (premature CAD patients) | [84] | [55] |
3. Correlation of Habits, Gender and COVID-19 with RAS Polymorphism
Still now, it is surprising that some COVID-19 patients are asymptomatic while others have much more severe disease outcomes. Moreover, viral respiratory infections are generally more harmful in children than in adults, but this appeared to be inverted in SARS-CoV-2 infection. It has been shown that some virus-related factors (viral load in the inoculum, the exposure duration and viral genomic mutations) can influence the severity and outcome of the disease [100][58] (Figure 2). Similarly, it quickly became obvious that several risk factors such as age, gender, the presence of comorbidities (such as smoking, immune status, diabetes, cardiovascular disease including hypertension, respiratory diseases and cancer) and the genetic background seem to control the manifestations and outcome of infection [41,45,101,102][59][60][61][62]. In addition, it has also drawn attention to vitamin D deficiency [103][63], as well as the ethnic differences, such as the black and South Asian ethnicities, and the lower socioeconomic statuses which are considered to increase risks [54][64]. Most mortalities occurred in the elderly and in men compared to women (4.7% vs. 2.8%). Moreover, the death of patients with no pre-existing conditions is approximately 10 times lower than in those with pre-existing ones. As in the 2003 SARS epidemic, hypertension has the most comorbidity frequency in non-survivors, in addition to cardiovascular diseases, obesity and diabetes, especially in smokers [54,90][64][65]. There is evidence that the RAS upregulation in the adipose tissue may lead to hypertension and insulin resistance in obese people. [37][66]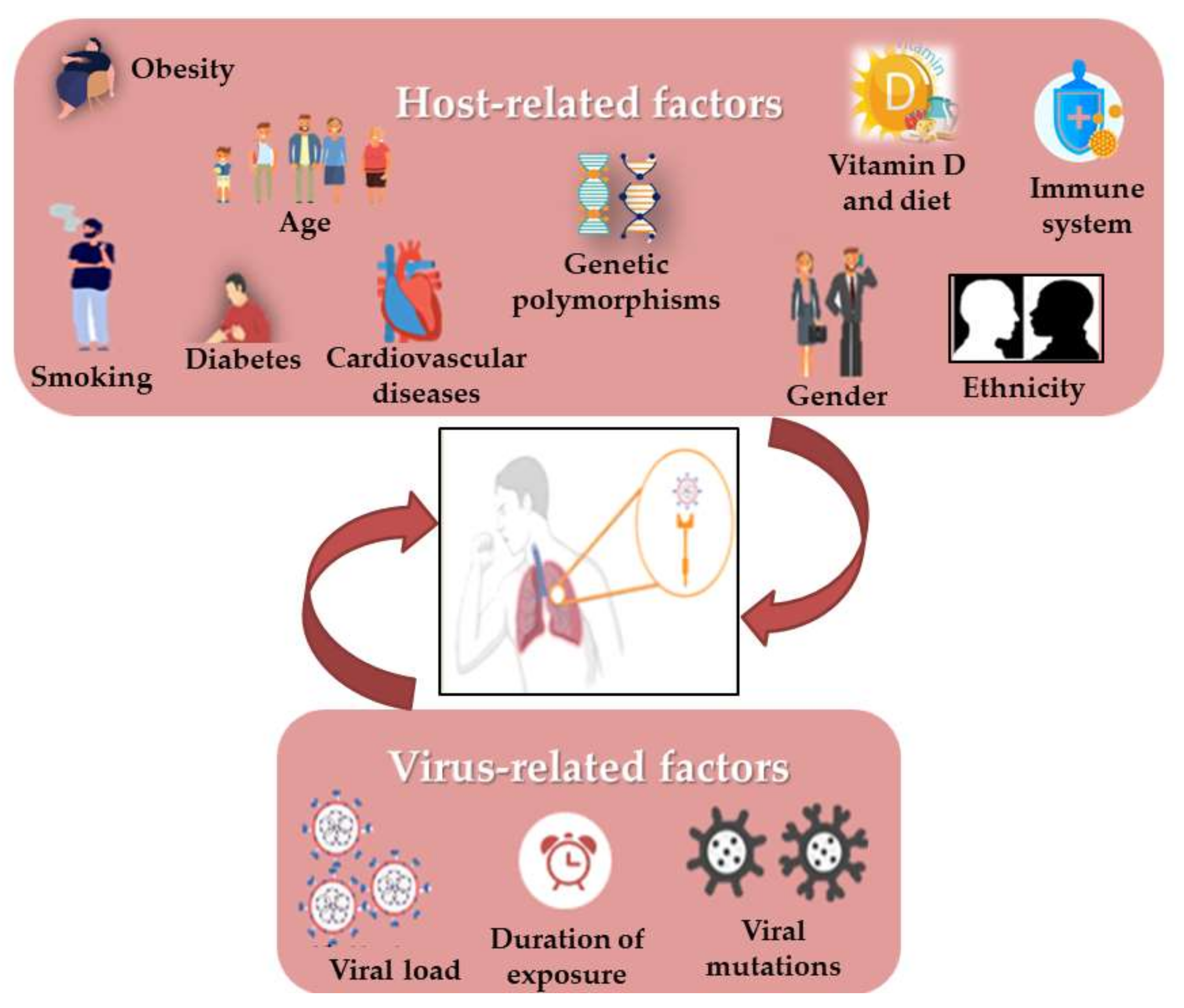
4. Conclusions
References
- World Health Organization. Available online: https://www.who.int/westernpacific/health-topics/detail/coronavirus (accessed on 13 September 2021).
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069.
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 475–486.
- Yuen, K.S.; Ye, Z.W.; Fung, S.Y.; Chan, C.P.; Jin, D.Y. SARS-CoV-2 and COVID-19: The most important research questions. Cell Biosci. 2020, 10.
- Bialek, S.; Boundy, E.; Bowen, V.; Chow, N.; Cohn, A.; Dowling, N.; Ellington, S.; Gierke, R.; Hall, A.; MacNeil, J. Severe Outcomes Among Patients with Coronavirus Disease 2019 (COVID-19)—United States. CDC 2021, 69, 343–346.
- Jirjees, F.; Saad, A.K.; Al Hano, Z.; Hatahet, T.; Al Obaidi, H.; Dallal Bashi, Y.H. COVID-19 Treatment Guidelines: Do They Really Reflect Best Medical Practices to Manage the Pandemic? Infect. Dis. Rep. 2021, 13, 29.
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease imhibitors: A review of molecular selectivity and toxicity. HIV/AIDS-Res. Palliat. Care 2014, 7, 95–104.
- Yu, B.; Li, C.; Chen, P.; Zhou, N.; Wang, L.; Li, J.; Jiang, H.; Wang, D.W. Low dose of hydroxychloroquine reduces fatality of critically ill patients with COVID-19. Sci. China Life Sci. 2020, 63, 1515–1521.
- Hadjadj, J.; Yatim, N.; Barnabel, L.; Corneau, A. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724.
- Miao, G.; Zhao, H.; Li, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2020, 56, 427–442.
- Liu, P.P.; Blet, A.; Smyth, D. The science underlying COVID-19, Implications for the cardiovascular system. Circulation 2020, 142, 68–78.
- Barré, J.; Sabatier, J.M.; Annweiler, C. Montelukast Drug May Improve COVID-19 Prognosis: A Review of Evidence. Front Pharmacol. 2020, 11, 1344–1349.
- Annweiler, C.; Papon, N.; Sabatier, J.M.; Barré, J. DAMPening Severe COVID-19 with Dexamethasone. Infect. Disord. Drug Targets 2021, 21.
- Vann, K.R.; Tencer, A.H.; Kutateladze, T.G. Inhibition of translation and immune responses by the virulence factor Nsp1 of SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 1–4.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020, 18, 271–280.
- Guo, X.; Chen, Z.; Xia, Y.; Lin, W.; Li, H. Investigation of the genetic variation in ACE2 on the structural recognition by the novel coronavirus (SARS-CoV-2). J. Transl. Med. 2020, 18, 321–330.
- Rysz, S.; Al-Saadi, J.; Sjöström, A.; Farm, M.; Jalde, F.C.; Plattén, M.; Eriksson, H.; Klein, M.; Vargas-Paris, R.; Nyrén, S. COVID-19 pathophysiology may be driven by an imbalance in the renin-angiotensin-aldosterone system. Nat. Commun. 2021, 12, 1–12.
- Perlot, T.; Penninger, J.M. ACE2–From the renin–angiotensin system to gut microbiota and malnutrition. Microbes Infect. 2013, 15, 866–873.
- Annweiler, C.; Cao, Z.; Wu, Y.; Faucon, F.; Mouhat, S.; Kovacic, H.; Sabatier, J.M. Counter-regulatory ‘Renin-Angiotensin’ System-based Candidate Drugs to Treat COVID-19 Diseases in SARS-CoV-2-infected Patients. Infect. Disord. Drug Targets 2020, 20, 407–408.
- Coto, E.; Avanzas, P.; Gómez, J. The Renin–Angiotensin–Aldosterone System and Coronavirus Disease 2019. Eur. Cardiol. Rev. 2021, 16, e07.
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20.
- Cao, Z.; Wu, Y.; Faucon, E.; Sabatier, J.M. SARS-CoV-2 & Covid-19: Key-Roles of the ‘Renin-Angiotensin’ System/Vitamin D Impacting Drug and Vaccine Developments. Infect. Disord. Drug Targets 2020, 20, 348–349.
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE2. Circ. Res. 2021, 128, 1323–1326.
- Bezabih, Y.M.; Bezabih, A.; Alamneh, E.; Peterson, G.M.; Bezabhe, W. Comparison of renin–angiotensin–aldosterone system inhibitors with other antihypertensives in association with coronavirus disease-19 clinical outcomes. BMC Infect. Dis. 2021, 21, 527.
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin–Angiotensin–Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659.
- Rafiullah, M. Can a Combination of AT1R Antagonist and Vitamin D Treat the Lung Complication of COVID-19? Am. J. Med Sci. 2020, 360, 338–341.
- Feng, Y.; Hans, C.; Mcllwain, E.; Varner, K.J.; Lazartigues, E. Angiotensin-Converting Enzyme 2 Over-Expression in the Central Nervous System Reduces Angiotensin-II- Mediated Cardiac Hypertrophy. PLoS ONE 2012, 7, e48910.
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116.
- Ghafil, F.; Hadi, N.R.; Mohammad, B.I.; Al-Aubaidy, H.A. Genetic Polymorphism of Angiotensin II Type 1 Receptors and Their Effect on the Clinical Outcome of Captopril Treatment in Arab Iraqi Patients with Acute Coronary Syndrome (Mid Euphrates). Ind. J. Clin. Biochem. 2019, 36, 81–87.
- Zhang, Y.H.; Zhang, Y.H.; Dong, X.F.; Hao, Q.Q.; Zhou, X.M.; Yu, Q.Y.; Li, S.Y.; Chen, X.; Tengbeh, A.F.; Dong, B.; et al. ACE2 and Ang-(1–7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm. Res. 2015, 64, 253–260.
- Hou, L.; Quan, X.; Li, X.; Su, X. Correlation between gene polymorphism in angiotensin II type 1 receptor and type 2 diabetes mellitus complicated by hypertension in a population of Inner Mongolia. BMC Med Genet. 2020, 21, 83.
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, 1377–1384.
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.Y.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370.
- Srivastava, A.; Bandopadhyay, A.; Das, D.; Pandey, R.; Singh, V.; Khanam, N.; Srivastava, N.; Singh, P.P.; Dubey, P.K.; Pathak, A.; et al. Genetic Association of ACE2 rs2285666 Polymorphism With COVID-19 Spatial Distribution in India. Front. Genet. 2020, 11, 1163.
- Cafiero, C.; Rosapepe, F.; Palmirotta, R.; Re, A.; Ottaiano, M.P.; Benincasa, G.; Perone, R.; Varriale, E.; D’Amato, G.; Cacciamani, A.; et al. Angiotensin System Polymorphisms’ in SARS-CoV-2 Positive Patients: Assessment Between Symptomatic and Asymptomatic Patients: A Pilot Study. Pharm. Pers. Med. 2021, 14, 621–629.
- Yamamoto, N.; Ariumi, Y.; Nishida, N.; Yamamoto, R.; Bauer, G.; Gojobori, T.; Shimotohno, K.; Mizokami, M. SARS-CoV-2 infections and COVID-19 mortalities strongly correlate with ACE1 I/D genotype. Gene 2020, 758, 144944.
- Devaux, C.A.; Pinault, L.; Osman, I.O.; Raoult, D. Can ACE2 Receptor Polymorphism Predict Species Susceptibility to SARS-CoV-2? Front. Public Health 2020, 8.
- Adamzik, M.; Frey, U.; Sixt, S.; Knemeyer, L.; Beiderlinden, M.; Peters, J.; Siffert, W. ACE I/D but not AGT (-6)A/G polymorphismis a risk factor for mortality in ARDS. Eur. Respir. J. 2007, 29, 482–488.
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W. Presenting Characteristics, Comorbidities, and OutcomesAmong 5700 Patients Hospitalized WithCOVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059.
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879.
- Hatami, N.; Ahi, S.; Fereydoni, M.; Sadeghinikoo, A.; Keshavarz, P.; Foroughian, M.; Osseini, A. Worldwide ACE (I/D) polymorphism may affect COVID-19 recovery rate: An ecological meta-regression. Endocrine 2020, 68, 479–484.
- I’ñiguez, M.; Pe´rez-Matute, P.; Villoslada-Blanco, P.; Recio-Fernandez, E.; Ezquerro-Pe´rez, D.; Alba, J.; Ferreira-Laso, M.L.; Oteo, J.A. ACE Gene Variants Rise the Risk of Severe COVID-19 in Patients with Hypertension, Dyslipidemia or Diabetes: A Spanish Pilot Study. Front. Endocrinol. 2021, 12.
- Verma, S.; Abbas, M.; Verma, S.; Khand, F.H.; Raza, S.T.; Siddiqi, Z.; Ahmada, I.; Mahdi, F. Impact of I/D polymorphism of angiotensin-converting enzyme 1 (ACE1) gene on the severity of COVID-19 patients. Infect. Genet. Evol. 2021, 91.
- Saab, Y.B.; Gard, P.R.; Overall, A.D.J. The geographic distribution of the ACE II genotype: A novel finding. Genet. Res. 2007, 89, 259–267.
- Mathew, J.; Basheeruddin, K.; Prabhaka, S. Differences in Frequency of the Deletion Polymorphism of the Angiotensin-Converting Enzyme Gene in Different Ethnic Groups. Angiology 2001, 52, 375–379.
- Marshall, R.P.; Webb, S.; Bellingan, G.J.; Montgomery, H.E.; Chaudhari, B.; McAnulty, R.J.; Humphries, S.E.; Hill, M.R.; Laurent, G.J. Angiotensin Converting Enzyme Insertion/Deletion Polymorphism Is Associated with Susceptibility and Outcome in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2002, 166, 646–650.
- Jerng, J.S.; Yu, C.J.; Wang, H.C.; Chen, K.Y.; Cheng, S.L.; Yang, P.C. Polymorphism of the angiotensin-converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit. Care Med. 2006, 34, 1001–1006.
- Calabrese, C.; Annunziata, A.; Coppola, A.; Pafundi, P.C.; Guarino, S.; Di Spirito, V.; Maddaloni, V.; Pepe, N.; Fiorentino, G. ACE Gene I/D Polymorphism and Acute Pulmonary Embolism in COVID19 Pneumonia: A Potential Predisposing Role. Front. Med. 2021, 7, 1136.
- Rani, B.; Kumar, A.; Bahl, A.; Sharma, R.; Prasad, R.; Khullar, M. Renin–angiotensin system gene polymorphisms as potential modifiers of hypertrophic and dilated cardiomyopathy phenotypes. Mol. Cell Biochem. 2017, 427, 1–11.
- Pouladi, N.; Abdolahi, S. Investigating the ACE2 polymorphisms in COVID- 19 susceptibility: An in silico analysis. Mol. Genet. Genomic. Med. 2021, 9.
- Imen, T.; Grissa, M.H.; Boubaker, H.; Beltaief, K.; Messous, S.; Tounsi, N.; Slimani, A.; Khouloud, C.; Bouida, W.; Boukef, R.; et al. AGT M235t polymorphism and heart failure in a cohort of Tunisian population: Diagnostic and prognostic value. Int. J. Clin. Exp. Med. 2015, 8, 16346–16351.
- Tran, T.T.; Mai, T.P.; Tran, H.C.B.; Le, L.H.G.; Vu, H.A.; Tran, T.K.; Hoang, S.V.; Chau, H.N.; Do, M.D. Association Between AGT M235T and Left Ventricular Mass in Vietnamese Patients Diagnosed with Essential Hypertension. Front. Cardiovasc. Med. 2021, 8, 16.
- Raygan, F.; Karimian, M.; Rezaeian, A.; Bahmani, B.; Behjati, M. Angiotensinogen-M235T as a risk factor for myocardial infarction in Asian populations: A genetic association study and a bioinformatics approach. Croat. Med. J. 2016, 57, 351–362.
- Pilbrow, A.P.; Palmer, B.R.; Frampton, C.M.; Yandle, T.G.; Troughton, R.W.; Campbell, E.; Skelton, L.; Lainchbury, J.G.; Richards, A.M.; Cameron, V.A. Angiotensinogen M235T and T174M Gene Polymorphisms in Combination Doubles the Risk of Mortality in Heart Failure. Hypertension 2007, 49, 322–327.
- Abd El-Aziz, T.A.; Mohamed, R.H.; Rezk, N.A. Association of angiotensin II type I and type II receptor genes polymorphisms with the presence of premature coronary disease and metabolic syndrome. Mol. Biol. Rep. 2014, 41, 1027–1033.
- Shahin, D.S.; Irshaid, Y.M.; Abu Saleh, A.S. The A1166C polymorphism of the AT1R gene is associated with an early onset of hypertension and high waist circumference in Jordanian males attending the Jordan University Hospital. Clin. Exp. Hypertens. 2014, 36, 333–339.
- Alavi-Shahri, J.; Behravan, J.; Hassany, M.; Tatari, F.; Kasaian, J.; Ganjali, R.; Tavallaie, S.; Sabouri, S.; Sahebkar, A.; Oladi, M.; et al. Association Between Angiotensin II Type 1 Receptor Gene Polymorphism and Metabolic Syndrome in a Young Female Iranian Population. Arch. Med. Res. 2010, 41, 343–349.
- Miller, D.S.; Kok, T.; Li, P. The virus inoculum volume influences outcome of influenza A infection in mice. Lab. Anim. 2013, 47, 74–77.
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163.
- Asselta, R.; Paraboschi, E.V.; Mantovani, A.; Stefano Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 2020, 12, 10087–10098.
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, Z.M.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk factors for severity and mortality in adult COVID-19 in patients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118.
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513.
- Crafaa, A.; Cannarellaa, R.; Condorellia, R.A.; Mongioìa, L.M.; Barbagalloa, F.; Aversab, A.; La Vigneraa, S.; Calogero, A.E. Influence of 25-hydroxy-cholecalciferol levels on SARS-CoV-2 infection and COVID-19 severity: A systematic review and meta-analysis. EClinicalMedicine 2021, 37, 100967.
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481.
- Hamet, P.; Pausova, Z.; Attaoua, R.; Hishmih, C.; Haloui, M.; Jean Shin, J.; Paus, T.; Abrahamowicz, M.; Gaudet, D.; Santucci, L.; et al. SARS-COV-2 receptor ACE2 gene is associated with hypertension and severity of COVID 19: Interaction with sex, obesity and smoking. Am. J. Hypertens. 2021, 34.
- Abdollahi, M.R.; Gaunt, T.R.; Syddall, H.E.; Cooper, C.; Phillips, D.I.; Ye, S.; Day, I.N.M. Angiotensin II type I receptor gene polymorphism: Anthropometric and metabolic syndrome traits. J. Med. Genet. 2005, 42, 396–401.