The 3D organoid model system represents a powerful tool for capturing the physiology of the normal or neoplastic esophagus. These 3D organoids are easily manipulatable, require little patient material, and are amenable to medium- or high-throughput screening. While no studies have yet leveraged the 3D organoid system to characterize the functional consequences of microbiome alterations in esophageal neoplasia, this system has been applied to other cancer types.
- 3D organoids
- esophageal adenocarcinoma
- esophageal squamous cell carcinoma
- Barrett’s esophagus
- microbiome
- dysbiosis
- barrier function
- host–pathogen interactions
1. Introduction
In this review, we will briefly summarize the relationship between the microbiome and esophageal neoplasia, discuss 3D organoid models of esophageal malignancies, highlight analogous GI models of host–pathogen interactions, and underscore the value of applying such models to esophageal disease.
ESCC is the predominant histological subtype of esophageal cancer worldwide, accounting for >90% of all esophageal cancers [27,28][1][2]. In recent years, there has been considerable progress documenting the genetic and epigenetic changes promoting ESCC initiation and development of esophageal cancer [29,30,31,32][3][4][5][6]. Despite these advancements, overall survival remains poor at approximately 20% [33][7]. Therefore, more work is needed to characterize both the intrinsic and extrinsic factors that promote ESCC tumorigenesis and to translate these findings into actionable therapeutic strategies.
Ultimately, the esophagus harbors a stable bacterial microbiome that is altered in esophageal neoplasia. Critically, whether these alterations have a causal role in promoting esophageal neoplasia is unclear. Few studies have addressed the functional consequences of alterations to these microbiomes on the identity and behavior of the underlying esophageal squamous epithelium. Such studies have utilized co-culture models of specific bacteria and esophageal epithelial cells in monolayer. While valuable, such models fail to recapitulate the dynamic esophageal squamous epithelium ( Section 1.1 ) and therefore offer an incomplete understanding of the impact of the microbiome on the underlying tissue. Faithful characterization of this impact will isolate the salient changes in these microbiomes as well as reveal novel therapeutic strategies to more effectively treat two devastating diseases.
In addition to ASC or cell line-based organoids, iPSC-based esophageal organoid models have recently been developed [77,78][8][9]. Esophageal progenitor cells (EPCs) can be generated through sequential specification of human pluripotent stem cells. EPCs can then be differentiated to recapitulate the normal development of the esophageal squamous epithelium. This system is generally used to study the development of the fetal esophagus, but organoids take several weeks to form, which is significantly slower than the formation of organoids from ASCs or cell lines. Together, there are several methods for forming 3D esophageal organoids, each with their strengths and weaknesses.
2. Organoid and Microbiome Co-Culture Models of GI Cancer-Relevant Processes
Elsewhere in the GI tract, co-culture models of 3D organoids and the gut microbiome have enabled the faithful characterization of the consequences of microbe–epithelial interactions [17,80,81][10][11][12]. These studies have focused on the effect of specific bacteria or bacterial metabolites on cancer-relevant processes in the gastric or intestinal epithelium, including proliferation, viability, inflammatory signaling, immunogenicity, genomic stability, and cell fate determination ( Table 1 ). To date, most studies have focused on the effect of Helicobacter pylori , a causative agent of gastric cancer, on gastric 3D organoids [82][13]. However, there is growing interest in modeling the interactions of the GI epithelia and other bacterial species using 3D organoids. We will highlight the GI cancer-relevant studies below and discuss how the 3D organoid system facilitates research in this clinically relevant and incompletely understood field.
| Tissue | Microbe | Classification | Product | Model | Host | Cancer-Associated Phenotype | Reference |
|---|---|---|---|---|---|---|---|
| Gastric | H. pylori | Pathogenic | Whole bacteria | Luminal microinjection | Human | Increased PD-L1 expression, increased survival | [8] |
| Increased inflammatory cytokine production (CXCL2, CXCL16, CXCL17, and CCL20), DC recruitment | [9] | ||||||
| Increased proliferation through c-Met signaling | [14] | ||||||
| Increased inflammatory cytokine production through the NF-κB pathway | [11] | ||||||
| Mouse | Increased proliferation through β-catenin signaling, mislocalization of Claudin-7 | [12] | |||||
| Human; Mouse | Increased CD44-dependent proliferation and EMT | [13] | |||||
| Intestinal | pks + E. coli | Pathogenic | Whole bacteria | Luminal microinjection | Human | Increased DNA damage and mutational burden | [15] |
| Mouse | Increased proliferation, decreased differentiation, increased chromosomal alterations, increased DNA mutational burden | [16] | |||||
| E. coli | Commensal | Whole bacteria | Luminal microinjection | Human | Increased proliferation (transient), enhanced barrier integrity through IL-6 and IL-8 signaling | [17] | |
| LPS | Supplemented into media | Mouse | Decreased proliferation, increased apoptosis through TLR4 signaling | [18] | |||
| Acinetobacter, Stenotrophomonas, and Delftia genera | Commensal | LPS | Supplemented into media | Mouse | Decreased proliferation, increased necroptosis, increased differentiation through TLR4 signaling | [19] | |
| L. reuteri D8 | Commensal | Whole bacteria, indole-3-aldehyde | Supplemented into media | Mouse | Increased proliferation, enhanced barrier integrity through IL-22 signaling | [20] | |
| Common commensal metabolites | Commensal | Gallic acid | Supplemented into media | Mouse | Increased WNT signaling, Increased proliferation, decreased differentiation in mutant p53 epithelial cells | [21] |
Unconstrained proliferation is a common feature of cancer cells. Recent studies have leveraged the 3D organoid platform to better characterize the influence of the microbiome on epithelial cell growth and have identified several different species that promote the proliferation of both gastric and intestinal epithelial organoids ( Table 1 ). An early study determined that microinjection of H. pylori into the lumen of human gastric organoids results in increased epithelial proliferation through c-Met signaling [83][15]. A similar study corroborated these findings, demonstrating that H. pylori microinjection into the lumen of murine-derived gastric organoids induced proliferation in a CagA- and β-catenin-dependent manner [84][16]. Further, H. pylori resulted in the mislocalization of claudin-7, a tight junction protein required to maintain mucosal epithelial integrity [84][16]. Further evidence from 3D organoid models suggests that H. pylori infection results in both increased proliferation of both patient- and murine-derived gastric organoids and increased epithelial –mesenchymal transition in a CD44-dependent manner [85][17]. Pretreatment of patient-derived organoids with a CD44 peptide inhibitor resulted in the loss of epithelial proliferation following exposure to H. pylori , demonstrating how findings from 3D co-culture models can reveal potential clinic targets for the treatment of microbiome-associated gastric cancers. Together, these studies demonstrate how 3D organoids can be utilized to characterize the molecular consequences of cancer-relevant microbe–epithelial interactions. Beyond H. pylori infection, recent evidence suggests that commensal microbiome metabolites can greatly influence the tumorgenicity and proliferative capacity of transformed epithelial tissue [86][18]. Intestinal tumor organoids derived from mice harboring oncogenic p53 mutations exhibit normal and balanced growth and differentiation in the absence of the microbiome through the disruption of the WNT pathway. However, treatment of these organoids with the bacterial metabolite gallic acid was sufficient to restore T-cell factor-mediated WNT signaling, increase organoid proliferative capacity, and result in a loss of organoid differentiation consistent with transformation. Removal of gallic acid from the culture medium reversed the transformed phenotype, highlighting the plasticity of these cells and presenting the intriguing possibility that modulation of the gut microbiota may be a potential therapeutic avenue for p53-mutated intestinal cancers. Highlighting the value of the 3D organoid system, the authors performed a coarse screen of the effect of many differential bacterial metabolites on intestinal tumor organoid growth and proliferation. This screen was possible because organoids capture the physiology of the original tissue and are easily treated and tracked. Ultimately, 3D organoids facilitated the discovery of a novel and highly cancer-relevant phenotype. In other contexts, 3D organoids have been utilized to demonstrate that bacterial products result in reduced proliferation and increased stem cell death. Treatment of murine intestinal crypt organoids with E. coli -derived endotoxin lipopolysaccharide (LPS) results in increased levels of the apoptotic marker cleaved caspase 3 and decreased levels of the proliferation marker PCNA [87][19]. LPS stimulation had no effect on Toll-like receptor 4 (TLR4) knockout mice. A similar study corroborated these results, demonstrating that LPS stimulation of murine intestinal organoids results in decreased proliferation, increased necroptosis (a programmed form of inflammatory cell death) of stem cells, and increased cell differentiation through a TLR4-dependent program [88][20]. This study is an elegant example of how the 3D organoid platform can be utilized to identify the molecular mechanisms and consequences of microbe–epithelial interactions. The authors isolated a crypt-specific core microbiota (CSCM) and hypothesized that this bacterial population affects epithelial generation. The authors first determined that the CSCM affected epithelial proliferation and survival in mice, and then employed the 3D organoid system to identify the salient molecular processes driving this change. The authors incubated organoids with sonicates and with purified LPS from four representative CSCM species ( S. maltophilia , A modestus , A. radioresistens , and D. tsuruhatensis ) and measured proliferation, death, and differentiation of epithelial cells. The authors determined that, while LPS from all CSCM species resulted in decreased organoid maturation, LPS from S. maltophilia specifically induced epithelial cell differentiation and RIPK3-dependent necroptosis of intestinal stem cells. The use of 3D organoids facilitated these studies by providing a physiologically-relevant platform to measure intestinal epithelial homeostasis using short (7 day) cultures that were easily scaled to include a variety of different bacterial byproducts. Together, these studies demonstrate that 3D GI organoids are a valuable platform for identifying the molecular mechanisms regulating epithelial proliferation and survival.
Inflammation is an enabling characteristic of cancer [89][21]. Recent studies have demonstrated that the co-culture of GI organoids and common GI microbes results in a strong inflammatory response ( Table 1 ). Microinjection of H. pylori into the lumen of gastric organoids results in a rapid (2 h) increase in NF-κB -regulated proinflammatory genes, including IL-8 [90][22]. Contradicting data from 2D cell lines, IL-8 expression in gastric organoids did not depend on bacterial cytotoxicity-associated gene pathogenicity island ( cag PAI) [90,91,92][22][23][24]. These data highlight how experiments performed in 3D organoids and 2D cell lines can produce different results. Organoid co-culture models have also revealed that the pro-inflammatory response to commensal bacterial metabolites can promote epithelial homeostasis. Co-culture of intestinal organoids and the commensal bacteria Lactobacillus reuteri ( L. reuteri ) D8 revealed that D8 metabolites stimulate IL-22 expression following intestinal injury, which accelerates epithelial proliferation and promotes barrier integrity [93][25]. Consistent with these data, microinjection of nonpathogenic E. coli into the lumen of intestinal organoids results in increased secretion of IL-6 and IL-8, a transient increase in proliferation, and improved epithelial barrier function [94][26]. These experiments demonstrate how organoid co-culture contextualizes the effects of the microbiome on the GI epithelium by faithfully recapitulating epithelial barrier function. Avoiding immune destruction is an emerging hallmark of cancer [89][21]. Recent evidence from microbiome and organoid co-culture has demonstrated that the microbiome can promote immune evasion ( Table 1 ). A co-culture model of H. pylori infection in patient-derived organoids and autologous patient cytotoxic T lymphocytes and dendritic cells (DCs) revealed that H. pylori induces programmed death-ligand 1 (PD-L1) expression through the Shh signaling pathway [95][27]. PD-L1 upregulation was rapid (within 48 h) and promoted epithelial cell survival. Treatment with an inhibitor of PD-L1 or programmed cell death protein 1 (PD-1) resulted in epithelial cell death, indicating that H. pylori -associated gastric tumors may be susceptible to immunotherapy. An additional study examined the co-culture of patient derived gastric organoids, luminally-microinjected H. pylori , and human monocyte-derived dendritic cells [96][28]. The authors demonstrated that H. pylori infection resulted in the recruitment of DCs to the gastric epithelia following the production of multiple chemokines, including CXCL2, CXCL16, CXCL17, and CCL20. These results indicate that the gastric epithelium can recruit DCs for immunosurveillance following H. pylori infection. Together, these data highlight how organoid co-culture models can be used to characterize important and targetable mechanisms underlying microbiome-associated GI cancers.
Loss of genomic stability is an enabling characteristic of cancer [89][21]. How bacteria may promote mutagenesis is unclear, in part due to the challenges performing long term co-culture experiments with human epithelial cells and microbiomes. To address this knowledge gap, a recent study performed a long term (5 months) co-culture through repeated microinjection of pathogenic polyketide synthetase ( pks ) + E. coli into healthy human intestinal organoids [97][29]. The authors demonstrated that pks + E. coli generate DNA damage and a distinct mutational signature that is commonly identified in colorectal cancer. Further, short-term infection of primary murine colon organoids with pks + E. coli results in phenotypes consistent with malignant transformation, including chromosomal aberrations, increased mutational burden, enhanced proliferation, and impaired differentiation [98][30]. Together, these findings leverage the 3D organoid platform to suggest that a pathogenic bacteria strain has a causal role in GI cancer transformation.
3. Discussion and Future Directions
3D organoids provide an intriguing platform for the study of epithelial–microbiome interactions for a variety of reasons. These organoids can be rapidly generated (<14 days) and passaged multiple times [71][31]. Additionally, the equipment and reagents required to culture 3D organoids are available in a modern molecular biology laboratory that performs 2D tissue culture [71][31]. Further, 3D organoids recapitulate the dynamic proliferation-differentiation gradient of the esophageal mucosa and are embedded in Matrigel, which simulates the basement matrix [71][31]. Therefore, this system is more physiologically relevant than 2D cell culture. Further, organoids can be established from patient samples or from isogenic mouse models. This versatility enables organoids to be used as a platform for personalized medicine or for targeted interrogation of the interaction of specific genes with the microenvironment [71][31]. Building on this versatility, 3D organoids are amenable to CRISPR-mediated or RNA interference (RNAi)-mediated genomic engineering and can be used for high-throughput screening in the presence of bacteria or bacterial metabolites added to the cell culture media [68][32]. Ultimately, 3D organoids represent a powerful tool for modeling epithelial–bacterial microbiome interactions in a physiologically relevant way.
However, there are limitations to 3D organoids as platforms for studying epithelial–microbiome interactions. There is a significant learning curve for generating 3D organoids from single cells [99][33]. Further, maintaining the oxygen gradient and/or anaerobic conditions required for the cultivation of specific aerobic/anaerobic bacterial species is challenging in the setting of tissue culture [81,99][12][33]. Additionally, microinjection of bacterial species into the organoid lumen is not well-suited to high-throughput screening. Further, the cell of origin for EAC and BE is controversial and may not be of esophageal origin, so co-culturing normal esophageal organoids with potentially pathogenic bacteria may be exploring early neoplastic changes in the wrong lineage [20][34]. Finally, reductionist approaches of co-culturing a single or a select few bacterial species with 3D organoids may occlude a common function of commensal bacteria: preventing the colonization of pathogenic bacteria [100][35]. Therefore, while the co-culture of 3D organoids and the bacterial microbiome is a promising and novel approach for studying host–microbiome interactions in esophageal neoplasia, there are limitations to this model system.
This approach can be applied to study the direct impact of the microbiome on esophageal neoplasia. Several specific bacterial phyla or species have been implicated as risk factors for ESD and ESCC progression, including increased levels of Fusobacteria and decreased levels of Actinobacteria [55,59][36][37]. Characterizing the effect of these and other phyla or species derived from normal, dysplastic, or ESCC tissue, as discussed in Section 2.2 and Section 2.3 on organoids, could provide insight into a potential role for bacteria in the pathobiology of ESCC initiation and development. Similar studies can address the effect of common microbial alterations in EAC and BE. Specifically, the co-culture of 3D organoids from BE or EAC patients with bacteria from the Campylobacter genera would enable insight into the mechanistic effects of a common alteration associated with BE. Further, altering the Streptococcus:Prevotella ratio in co-culture models of BE organoids could identify the molecular changes that accompany a common microbial alteration [62,63][38][39]. Finally, co-culture of EAC-derived organoids with Veillonella could provide evidence that a controversial alteration is or is not contributing to EAC pathobiology [57,62][40][38]. Each proposed experiment would clarify the consequences of common microbial alterations in esophageal neoplasia.
Ultimately, the microbiome is an emerging co-factor in esophageal health and disease. While many studies have documented common components of normal and transformed esophageal epithelial cells, the current understanding of the role of the bacterial microbiome in esophageal homeostasis remains largely descriptive. The co-culture of 3D organoids with both individual bacterial species or the bacterial microbiome isolated from patients will enable functional annotation of these changes ( Figure 1 ).
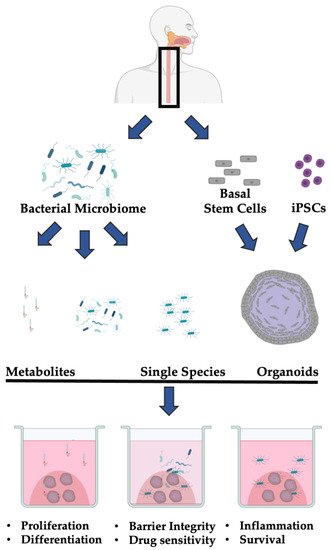
4. Conclusions
The microbiome is an emergent co-factor in the pathobiology of esophageal neoplasia [41]. Within the past two decades, several studies have determined that the composition of the esophageal or oral microbiome is altered in ESCC and EAC as well as their respective precursor lesions ESD and BE. These alterations may have both prognostic and therapeutic value; however, more work is needed to characterize the functional consequences of these changes. The 3D organoid model system represents a powerful tool for capturing the physiology of the normal or neoplastic esophagus. These 3D organoids are easily manipulatable, require little patient material, and are amenable to medium- or high-throughput screening. While no studies have yet leveraged the 3D organoid system to characterize the functional consequences of microbiome alterations in esophageal neoplasia, this system has been applied to other cancer types. Elsewhere in the GI tract, co-culture models of gastric or intestinal 3D organoids have enabled mechanistic insights into how the bacterial microbiome can promote cancer-specific processes such as proliferation, inflammation, immune escape, and mutagenicity. These insights have provided potential therapeutic targets. Therefore, there is growing interest in applying 3D organoid technology to unravel the mechanistic consequences of epithelial–bacterial microbiome interactions in esophageal neoplasia. Additionally, 3D organoid esophageal organoids can be used to identify the functional consequences of epithelial interactions with other elements of the microbiome, including viruses such as HPV. Further, by expanding co-culture models of esophageal organoids with the microbiome and with other stromal or immune cell elements, researchers can better recapitulate the native environment of the human esophagus. This platform would be ideal for personalized medicine. Ultimately, the co-culture of esophageal organoids and the bacterial microbiome is an untapped platform with the potential to provide actionable insight into the pathobiology of a leading cause of cancer worldwide.
References
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509.
- Rustgi, A.K. Esophageal Cancers and Model Systems. Trans. Am. Clin. Climatol. Assoc. 2019, 130, 266–271.
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175.
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6.
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e6.
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30.
- Zhang, Y.; Yang, Y.; Jiang, M.; Huang, S.X.; Zhang, W.; Al Alam, D.; Danopoulos, S.; Mori, M.; Chen, Y.W.; Balasubramanian, R.; et al. 3D Modeling of Esophageal Development using Human PSC-Derived Basal Progenitors Reveals a Critical Role for Notch Signaling. Cell Stem Cell 2018, 23, 516–529.e5.
- Trisno, S.L.; Philo, K.E.D.; McCracken, K.W.; Catá, E.M.; Ruiz-Torres, S.; Rankin, S.A.; Han, L.; Nasr, T.; Chaturvedi, P.; Rothenberg, M.E.; et al. Esophageal Organoids from Human Pluripotent Stem Cells Delineate Sox2 Functions during Esophageal Specification. Cell Stem Cell 2018, 23, 501–515.e7.
- Min, S.; Kim, S.; Cho, S.W. Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp. Mol. Med. 2020, 52, 227–237.
- Puschhof, J.; Pleguezuelos-Manzano, C.; Martinez-Silgado, A.; Akkerman, N.; Saftien, A.; Boot, C.; de Waal, A.; Beumer, J.; Dutta, D.; Heo, I.; et al. Intestinal organoid cocultures with microbes. Nat. Protoc. 2021, 16, 4633–4649.
- Puschhof, J.; Pleguezuelos-Manzano, C.; Clevers, H. Organoids and organs-on-chips: Insights into human gut-microbe interactions. Cell Host Microbe 2021, 29, 867–878.
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78.
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664.
- McCracken, K.W.; Catá, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404.
- Wroblewski, L.E.; Piazuelo, M.B.; Chaturvedi, R.; Schumacher, M.; Aihara, E.; Feng, R.; Noto, J.M.; Delgado, A.; Israel, D.A.; Zavros, Y.; et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2015, 64, 720–730.
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M., Jr.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 Plays a Functional Role in Helicobacter pylori-induced Epithelial Cell Proliferation. PLoS Pathog. 2015, 11, e1004663.
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138.
- Neal, M.D.; Sodhi, C.P.; Jia, H.; Dyer, M.; Egan, C.E.; Yazji, I.; Good, M.; Afrazi, A.; Marino, R.; Slagle, D.; et al. Toll-like Receptor 4 Is Expressed on Intestinal Stem Cells and Regulates Their Proliferation and Apoptosis via the p53 Up-regulated Modulator of Apoptosis. J. Biol. Chem. 2012, 287, 37296–37308.
- Naito, T.; Mulet, C.; De Castro, C.; Molinaro, A.; Saffarian, A.; Nigro, G.; Bérard, M.; Clerc, M.; Pedersen, A.B.; Sansonetti, P.J.; et al. Lipopolysaccharide from crypt-specific core microbiota modulates the colonic epithelial proliferation-to-differentiation balance. MBio 2017, 8.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Bartfeld, S.; Bayram, T.; Van De Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015, 148, 126–136.e6.
- Bartfeld, S.; Hess, S.; Bauer, B.; Machuy, N.; Ogilvie, L.A.; Schuchhardt, J.; Meyer, T.F. High-throughput and single-cell imaging of NF-κB oscillations using monoclonal cell lines. BMC Cell Biol. 2010, 11.
- Ferrero, R.L. Innate immune recognition of the extracellular mucosal pathogen, Helicobacter pylori. Mol. Immunol. 2005, 42, 879–885.
- Hou, Q.; Ye, L.; Liu, H.; Huang, L.; Yang, Q.; Turner, J.; Yu, Q. Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2018, 25, 1657–1670.
- Hill, D.R.; Huang, S.; Nagy, M.S.; Yadagiri, V.K.; Fields, C.; Mukherjee, D.; Bons, B.; Dedhia, P.H.; Chin, A.M.; Tsai, Y.H.; et al. Bacterial colonization stimulates a complex physiological response in the immature human intestinal epithelium. Elife 2017, 6.
- Holokai, L.; Chakrabarti, J.; Broda, T.; Chang, J.; Hawkins, J.A.; Sundaram, N.; Wroblewski, L.E.; Peek, R.M.; Wang, J.; Helmrath, M.; et al. Increased Programmed Death-Ligand 1 is an Early Epithelial Cell Response to Helicobacter pylori Infection. PLoS Pathog. 2019, 15, 1–30.
- Sebrell, T.A.; Hashimi, M.; Sidar, B.; Wilkinson, R.A.; Kirpotina, L.; Quinn, M.T.; Malkoç, Z.; Taylor, P.J.; Wilking, J.N.; Bimczok, D. A Novel Gastric Spheroid Co-culture Model Reveals Chemokine-Dependent Recruitment of Human Dendritic Cells to the Gastric Epithelium. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 157–171.e3.
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks + E. coli. Nature 2020, 580, 269–273.
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat. Commun. 2021, 12.
- Nakagawa, H.; Kasagi, Y.; Karakasheva, T.A.; Hara, T.; Aaron, B.; Shimonosono, M.; Kijima, T.; Giroux, V.; Bailey, D.; Wilkins, B.; et al. Modeling Epithelial Homeostasis and Reactive Epithelial Changes in Human and Murine Three-Dimensional Esophageal Organoids. Curr. Protoc. Stem Cell Biol. 2020, 52, e106.
- Sachdeva, U.M.; Shimonosono, M.; Flashner, S.; Cruz-Acuña, R.; Gabre, J.T.; Nakagawa, H. Understanding the cellular origin and progression of esophageal cancer using esophageal organoids. Cancer Lett. 2021, 509, 39–52.
- Luca, F.; Kupfer, S.S.; Knights, D.; Khoruts, A.; Blekhman, R. Functional Genomics of Host-Microbiome Interactions in Humans. Trends Genet. 2018, 34, 30–40.
- Hayakawa, Y.; Nakagawa, H.; Rustgi, A.K.; Que, J.; Wang, T.C. Stem cells and origins of cancer in the upper gastrointestinal tract. Stem Cell 2021, 28, 1343–1361.
- Goto, Y. Commensal bacteria prevent pathogenic bacterial infection by inducing of activation of host immune system. Nippon Saikingaku Zasshi 2020, 75, 185–194.
- Li, D.; He, R.; Hou, G.; Ming, W.; Fan, T.; Chen, L.; Zhang, L.; Jiang, W.; Wang, W.; Lu, Z.; et al. Characterization of the Esophageal Microbiota and Prediction of the Metabolic Pathways Involved in Esophageal Cancer. Front. Cell. Infect. Microbiol. 2020, 10, 268.
- Shao, D.; Vogtmann, E.; Liu, A.; Qin, J.; Chen, W.; Abnet, C.C.; Wei, W. Microbial characterization of esophageal squamous cell carcinoma and gastric cardia adenocarcinoma from a high-risk region of China. Cancer 2019, 125, 3993–4002.
- Lopetuso, L.R.; Severgnini, M.; Pecere, S.; Ponziani, F.R.; Boskoski, I.; Larghi, A.; Quaranta, G.; Masucci, L.; Ianiro, G.; Camboni, T.; et al. Esophageal microbiome signature in patients with Barrett’s esophagus and esophageal adenocarcinoma. PLoS ONE 2020, 15, e0231789.
- Deshpande, N.P.; Riordan, S.M.; Castaño-Rodríguez, N.; Wilkins, M.R.; Kaakoush, N.O. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome 2018, 6, 1–14.
- Elliott, D.R.F.; Walker, A.W.; O’Donovan, M.; Parkhill, J.; Fitzgerald, R.C. A non-endoscopic device to sample the oesophageal microbiota: A case-control study. Lancet Gastroenterol. Hepatol. 2017, 2, 32–42.
- Yano, Y.; Etemadi, A.; Abnet, C.C. Microbiome and Cancers of the Esophagus: A Review. Microorganisms 2021, 9, 1764.