Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Elena Tamagno and Version 3 by Jessie Wu.
The pathogenesis of Alzheimer's disease (AD) involves beta amyloid accumulation known to induce synaptic dysfunction and neurodegenration. The brain's vulnerability to oxidative stress (OS) is considered a crucial detrimental factor in AD. OS and beta amyloid are linked each other because beta amyloid induces OS and, in turn, OS induces beta amyloid accumulation. Evidence indicates that a gradual oxidative damage accumulation precedes and results in the appearance of pathological AD symptoms. Moreover, OS plays a crucial role in the pathogenesis of many risk factors for AD. This neurodegenerative disorder begins many years before symptoms, thus antioxidant treatment can represent a good therapeutic target fot attacking the disease,
- Alzheimer’s disease
- oxidative stress
1. Introduction
Alzheimer’s disease (AD) is considered the leading cause of dementia and is becoming one of the most expensive and deadly diseases of our time [1]. Thus, it is estimated that 50 million people worldwide endure dementia, and this number is set to rise to 152 million in 2050 [2][3]. Moreover, Alzheimer’s patients need specialized and expensive care, the annual cost of treatment worldwide is around a trillion US dollars, and it is predicted that this cost will significantly increase by 2030 [4].
The pathophysiology of the disease is complex and multifactorial and certainly not entirely known [3]. There are two markers of the disease. One is β amyloid (Aβ), which accumulates abnormally in AD brain tissues and forms extracellular plaques known to induce synaptic alterations and neurodegeneration [5][6]. The other is Tau protein, which forms intracellular neurofibrillary tangles that are also responsible for neurodegeneration [7][8].
AD is traditionally divided into two forms: early and late onset forms. The early onset form is closely associated with mutations on three genes: the genes that encode for the amyloid precursor (APP) and for presenilin 1 and presenilin 2, which represent the catalytic core of γ-secretase, a key enzyme for the production of Aβ [9]. The late onset form is caused by complex interactions between genetic and environmental factors [10][11]. APOE is the gene that was first associated with the development of the late form [12] compared to other predisposing genes that have been described. These include genes involved in neuroinflammation (such as TREM2, TYROBP, and CD33) [13][14][15], memory (CR1, PICALM, and BIN1) [16][17][18], and lipid metabolism (ABCA7 and CLU) [19][20].
Thus, although there are numerous factors associated with the development of the disease, Aβ represents one that is the most closely related to its pathogenesis. Aβ is composed of polypeptides of various lengths, and most of those found in the plaques are 40 and 42 amino acids long [6]. Aβ 40 is the most abundant form and accounts for 90% of the amyloid present in the plaques; however, the longer form (Aβ 42) is predominant in the initial phases and aggregates faster than Aβ 40 [21]. Aβ is produced from the amyloid precursor protein (APP). APP is an integral membrane protein with a large, extracellular N-terminus and a shorter, cytoplasmic C-terminus [22][23]. The amyloidogenic processing of APP involves two sequential cleavages operated by the β-secretases and γ-secretases. The β-secretase (BACE1) cleaves APP, generating an extracellular soluble fragment called sβAPP and an intracellular C-terminal end termed C99 [24]. C99 is further cleaved, within the membrane, by the γ-secretase [25]. The γ-cleavage produces Aβ fragments of different lengths, and these are predominantly Aβ 40 and Aβ 42 [25].
2. Aβ vs. Oxidative stress
There are numerous mechanisms described in the literature by which beta amyloid (Aβ) mediates oxidative damage (Figure 1).
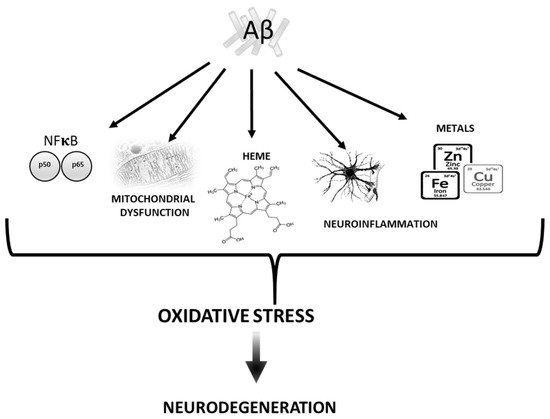
Figure 1. Diagram sketching mechanisms by which Aβ induces oxidative stress.
2.1. Mitochondria
Aβ interferes with the normal mitochondrial activity, causing dysfunction that results in oxidative stress [26]. Thus, neurons are cells that require high energy levels to perform numerous functions, such as the generation of action potentials, nerve transmission, and axonal transport [27].
The alteration of oxidative phosphorylation (OXPHOS) involves a reduction in the efficiency to transfer electrons, which in turn results in an increase in ROS production predominantly at level of complex I and complex III. These ROSs generated at the chain level unfold their damaging action mainly on mitochondrial macromolecules [28]. The peptide Aβ not only promotes the generation of ROS at the level of the mitochondria, but inhibits, at the same time, ROS removal. In fact, it has been shown that Aβ is able to inhibit mitochondrial superoxide dismutase (MnSOD), the enzyme most involved in the detoxification of the anion superoxide and protection from peroxidative damage [29][30][29,30]. Aβ is also capable of binding and inhibiting mitochondrial alcohol dehydrogenase known as ABAD (Aβ binding alcoholdehydrogenase). ABAD has a protective role, being responsible for the detoxification from aldehydes such as 4-hydroxinonenal. The interaction between the Aβ peptide and ABAD compromises the detoxification process for which the enzyme is responsible for, causing lipid peroxidation, ROS generation, and mitochondrial dysfunction [31].
Furthermore, from the direct impact of Aβ on mitochondria, some authors also demonstrated that mitochondrial DNA is altered in elderly and AD patients [32]. Various factors can influence mitochondrial activity, contributing to AD progression. [33]. In brain tissue of AD cases, there is a downregulation of genes in mitochondrial complex I of the OXPHOS [34], and OS is implicated in mtDNA damage [35].
2.2. Transition Metals
Another mechanism found dysregulated in AD is the homeostasis of metals such as iron (Fe), copper (Cu), and zinc (Zn). The blood–brain barrier tightly regulates the concentration of these metals, but their levels significantly increased in AD patients [36][37][36,37]. Studies have shown that Aβ plaques contain traces of these metals [38], and Aβ can reduce Fe (III) or Cu (II) to induce hydrogen peroxide (H2O2) production, contributing to OS in AD [39]. Moreover, some studies suggested that these metals can increase Aβ polymerization; thus, neuroblastoma cells treated with Fe3+ caused an increase in BACE1 activity that, in turn, promotes Aβ production [40]. Moreover, zinc is also abundant in amyloid plaques, suggesting its role in AD. Zinc interacts with Aβ protein, and this interaction can be prevented by chelators [41][42][41,42]. ZnAβ oligomers mediate stronger toxicity than amyloid-beta derived diffusible ligands (ADDLs) by cell viability assays [43]. The ex vivo study showed that ZnAβ oligomers inhibited hippocampal LTP in a transgenic mouse model also through the production of ROS [44].
2.3. Heme
Another important iron containing molecule that has been implicated in the pathogenesis of AD is heme. Heme is an essential molecule in various physiological and pathological mechanisms [45]. It has been suggested that AD patients had lower hemoglobin levels and smaller cell volume with respect to normal aging controls [46]. Complexes II, III, and IV of the electron transport chain require heme to assembly cytochromes needed to function [47]. Perturbation in heme metabolism can cause oxidative stress and cell death [48]. Sankar and collaborators found that heme can mitigate the Aβ 42-mediated neuroinflammation activated by astrocytes [49]. Heme can also bind to Aβ, and this complex is known to have peroxidase activity able to oxidize serotonin and DOPA and providing an intriguing link between heme and oxidative stress in AD [50]. Thus, heme deficiency is followed by formation of APP dimers and loss of complex IV of the electron transport chain, and this event causes OS [51]. This finding suggested that iron accumulation observed in AD could be strictly linked to heme deficiency [51][52].
2.4. Neuroinflammation
Another crucial mechanism through which the presence of Aβ induced oxidative stress is neuroinflammation [52][53]. Neuroinflammation is considered as an immunological response characterized by the activation of glial cells and the production of inflammatory mediators [53][54]. Numerous studies revealed a strong correlation between neuroinflammation and AD pathology [54][55][56][55,56,]; thus, inflammatory cytokines have been reported to increase in the progression of mild cognitive impairment to overt AD [57]. A microarray study of Cribbs and collaborators performed on young, aged, and AD cases demonstrated an upregulation of the innate immune response in aging brains and a slight increase in related genes [58], suggesting that inflammation has a role in the preclinical stages of AD. In this context, microglia play a leading role in neuroinflammation [53][59]. Aβ can bind different microglial receptors, resulting in the production not only of inflammatory cytokines and chemokines [58][60] but also of a large amount of oxygen free radicals (•OH and O₂•) [59][61], nitric oxide (•NO) [60][62], and tumour necrosis factor (TNF) α [61][63]. The NLR family pyrin domain containing 3 (NLRP3) inflammasome is a recently found cytoplasmic protein complex involved in neuroinflammation and innate immune response [62][64]. Recent studies demonstrated that Aβ induce NLRP3 activation in microglia and astrocytes. This event results in the production of caspase 1 and induces the release of cytokines such as IL1β and IL-18, resulting in irreversible damage. On the other hand, the inhibition of NLRP3 inflammasome inhibits Aβ deposition and had a neuroprotective effect in a transgenic AD mouse model [63][64][65][66][65,66]. Thus, all these findings suggest that Aβ is a crucial factor in AD associated inflammation and oxidative stress.
2.5. NF-kB Pathway
The NF-kB family also has an important role in modulating oxidative stress. Thus, evidence from in vitro studies showed increased oxidative stress-mediated by NF-kB in response to neurons exposed to Aβ; this increased oxidative stress resulted in the accumulation of lipid peroxides and neurodegeneration [67]. Moreover, many studies confirm that Aβ peptides stimulate NF-kB gene expression and its nuclear translocation [68]
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70.
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res Ther. 2016, 8, 39. Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R. Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer's disease: the path to 2025. Alzheimers Res Ther. 2016, 8, 39.
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590.
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 2019, 179, 312–339.
- Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030.
- Yu, H.; Wu, J. Amyloid-β: A double agent in Alzheimer’s disease? Biomed. Pharm. 2021, 139, 111575. Yu, H.; Wu, J. Amyloid-β: A double agent in Alzheimer’s disease? Biomed Pharm. 2021, 139, 111575.
- Ji, C.; Sigurdsson, E.M. Current Status of Clinical Trials on Tau Immunotherapies. Drugs 2021, 8, 1135–1152. Ji, C.; Sigurdsson, E.M. Current Status of Clinical Trials on Tau Immunotherapies. Drugs 2021, 8, Epub ahead of print.
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37. Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37.
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281.
- Haaksma, M.L.; Vilela, L.R.; Marengoni, A.; Calderón-Larrañaga, A.; Leoutsakos, J.S.; Olde Rikkert, M.G.M.; Melis, R.J.F. Comorbidity and progression of late onset Alzheimer’s disease: A systematic review. PLoS ONE 2017, 12, e0177044. Haaksma, M.L.; Vilela, L.R.; Marengoni, A.; Calderón-Larrañaga, A.; Leoutsakos, J.S.; Olde Rikkert, M.G.M.; Melis, R.J.F. Comorbidity and progression of late onset Alzheimer’s disease: A systematic review. PLoS ONE. 2017, 12, e0177044.
- Lemche, E. Early Life Stress and Epigenetics in Late-onset Alzheimer’s Dementia: A Systematic Review. Curr. Genom. 2018, 19, 522–602. Lemche, E. Early Life Stress and Epigenetics in Late-onset Alzheimer’s Dementia: A Systematic Review. Curr. Genom. 2018, 19, 522–602.
- Zhou, X.; Fu, A.K.; Ip, N.Y. APOE signaling in neurodegenerative diseases: An integrative approach targeting APOE coding and noncoding variants for disease intervention. Curr. Opin. Neurobiol. 2021, 69, 58–67. Zhou, X.; Fu, A.K.; Ip, N.Y. APOE signaling in neurodegenerative diseases: An integrative approach targeting APOE coding and noncoding variants for disease intervention. Curr. Opin. Neurobiol. 2021, 69, 58–67.
- Guglielmotto, M.; Repetto, I.E.; Monteleone, D.; Vasciaveo, V.; Franchino, C.; Rinaldi, S.; Tabaton, M.; Tamagno, E. Stroke and Amyloid-β Downregulate TREM-2 and Uch-L1 Expression that Synergistically Promote the Inflammatory Response. J. Alzheimers Dis. 2019, 71, 907–920. Guglielmotto, M.; Repetto, I.E.; Monteleone, D.; Vasciaveo, V.; Franchino, C.; Rinaldi, S.; Tabaton, M.; Tamagno, E. Stroke and Amyloid-β Downregulate TREM-2 and Uch-L1 Expression that Synergistically Promote the Inflammatory Response. J. Alzheimers Dis. 2019, 71, 907–920.
- Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J.; et al. Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s disease. J. Neurosci. Res. 2021, 99, 1704–1721. Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J.; et al. Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s disease. J. Neurosci. Res. 2021, 99, 1704–1721.
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ1-42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 19. Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ1-42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 19.
- Mitsumori, R.; Sakaguchi, K.; Shigemizu, D.; Mori, T.; Akiyama, S.; Ozaki, K.; Niida, S.; Shimoda, N. Lower DNA methylation levels in CpG island shores of CR1, CLU, and PICALM in the blood of Japanese Alzheimer’s disease patients. PLoS ONE 2020, 15, e0239196. Mitsumori, R.; Sakaguchi, K.; Shigemizu, D.; Mori, T.; Akiyama, S.; Ozaki, K.; Niida, S.; Shimoda, N. Lower DNA methylation levels in CpG island shores of CR1, CLU, and PICALM in the blood of Japanese Alzheimer’s disease patients. PLoS ONE 2020, 15, e0239196.
- Xu, W.; Tan, C.C.; Cao, X.P.; Tan, L. Alzheimer’s Disease Neuroimaging Initiative. Association of Alzheimer’s disease risk variants on the PICALM gene with PICALM expression, core biomarkers, and feature neurodegeneration. Aging 2020, 12, 21202–21219. Xu, W.; Tan, C.C.; Cao, X.P.; Tan, L. Alzheimer’s Disease Neuroimaging Initiative. Association of Alzheimer’s disease risk variants on the PICALM gene with PICALM expression, core biomarkers, and feature neurodegeneration. Aging 2020, 12, 21202–21219.
- Franzmeier, N.; Ossenkoppele, R.; Brendel, M.; Rubinski, A.; Smith, R.; Kumar, A.; Mattsson-Carlgren, N.; Strandberg, O.; Duerin, M.; Buerger, K.; et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI)* and the Swedish BioFINDER study. The BIN1 rs744373 Alzheimer’s disease risk SNP is associated with faster Aβ-associated tau accumulation and cognitive decline. Alzheimers Dement. 2021. in print. Franzmeier, N.; Ossenkoppele, R.; Brendel, M.; Rubinski, A.; Smith, R.; Kumar, A.; Mattsson-Carlgren, N.; Strandberg, O.; Duerin, M.; Buerger. K.; Dichgans, M.; et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI)* and the Swedish BioFINDER study. The BIN1 rs744373 Alzheimer’s disease risk SNP is associated with faster Aβ-associated tau accumulation and cognitive decline. Alzheimers Dement. 2021, Epub ahead of print.
- Dib, S.; Pahnke, J.; Gosselet, F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4603. Dib. S.; Pahnke, J.; Gosselet, F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4603.
- An, N.; Fu, Y.; Shi, J.; Guo, H.N.; Yang, Z.W.; Li, Y.C.; Li, S.; Wang, Y.; Yao, Z.J.; Hu, B. Alzheimer’s Disease Neuroimaging Initiative. Synergistic Effects of APOE and CLU May Increase the Risk of Alzheimer’s Disease: Acceleration of Atrophy in the Volumes and Shapes of the Hippocampus and Amygdala. J. Alzheimers Dis. 2021, 80, 1311–1327. An, N.; Fu, Y.; Shi, J.; Guo, H.N.; Yang, Z.W.; Li, Y.C.; Li, S.; Wang, Y.; Yao, Z.J.; Hu, B. Alzheimer’s Disease Neuroimaging Initiative. Synergistic Effects of APOE and CLU May Increase the Risk of Alzheimer’s Disease: Acceleration of Atrophy in the Volumes and Shapes of the Hippocampus and Amygdala. J. Alzheimers Dis. 2021, 80, 1311–1327.
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976-84.
- Nguyen, K.V. The human β-amyloid precursor protein: Biomolecular and epigenetic aspects. Biomol. Concepts. 2015, 6, 11–32. Nguyen, K.V. The human β-amyloid precursor protein: Biomolecular and epigenetic aspects. Biomol. Concepts. 2015, 6, 11–32.
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull 2017, 133, 71–79. Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull 2017, 133, 71–79.
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross , S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741.
- Huse, J.T.; Doms, R.W. Closing in on the amyloid cascade: Recent insights into the cell biology of Alzheimer’s disease. Mol. Neurobiol. 2000, 22, 81–98. Huse, J.T.; Doms, R.W. Closing in on the amyloid cascade: Recent insights into the cell biology of Alzheimer’s disease. Mol. Neurobiol. 2000, 22, 81–98.
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253.
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi Di Carlo, M. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid. Med. Cell Longev. 2014, 2014, 780179. Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi Di Carlo, M. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid. Med. Cell Longev. 2014, 2014, 780179.
- Desler, C.; Lillenes, M.S.; Tønjum, T.; Rasmussen, L.J. The Role of Mitochondrial Dysfunction in the Progression of Alzheimer’s Disease. Curr. Med. Chem. 2018, 25, 5578–5587. Desler, C.; Lillenes, M.S.; Tønjum, T.; Rasmussen, L.J. The Role of Mitochondrial Dysfunction in the Progression of Alzheimer’s Disease. Curr. Med. Chem. 2018, 25, 5578–5587.
- Anantharaman, M.; Tangpong, J.; Keller, J.N.; Murphy, M.P.; Markesbery, W.R.; Kiningham, K.K.; St Clair, D.K. Beta-amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol. 2006, 168, 1608–1618. Anantharaman, M.; Tangpong, J.; Keller, J.N.; Murphy, M.P.; Markesbery, W.R.; Kiningham. K.K.; St Clair, D.K. Beta-amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol. 2006, 168, 1608–1618.
- Sen, A.; Nelson, T.J.; Alkon, D.L.; Hongpaisan, J. Loss in PKC Epsilon Causes Downregulation of MnSOD and BDNF Expression in Neurons of Alzheimer’s Disease Hippocampus. J. Alzheimers Dis. 2018, 63, 1173–1189. Sen, A.; Nelson, T.J.; Alkon, D.L.; Hongpaisan, J. Loss in PKC Epsilon Causes Downregulation of MnSOD and BDNF Expression in Neurons of Alzheimer’s Disease Hippocampus. J. Alzheimers Dis. 2018, 63, 1173–1189.
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320.
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195.
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim Biophys Acta. 2014, 1842, 1219–1231.
- Detaille, D.; Pasdois, P.; Sémont, A.; Dos Santos, P.; Diolez, P. An old medicine as a new drug to prevent mitochondrial complex I from producing oxygen radicals. PLoS ONE 2019, 14, e0216385. Detaille, D.; Pasdois, P.; Sémont, A.; Dos Santos, P.; Diolez, P. An old medicine as a new drug to prevent mitochondrial complex I from producing oxygen radicals. PLoS ONE 2019, 14, e0216385.
- Bennett, J.P. Medical hypothesis: Neurodegenerative diseases arise from oxidative damage to electron tunneling proteins in mitochondria. Med. Hypotheses 2019, 127, 1–4. Bennett, J.P. Medical hypothesis: Neurodegenerative diseases arise from oxidative damage to electron tunneling proteins in mitochondria. Med. Hypotheses 2019, 127, 1–4.
- Qi, Z.; Liu, K.J. The interaction of zinc and the blood-brain barrier under physiological and ischemic conditions. Toxicol. Appl. Pharmacol. 2019, 364, 114–119. Qi, Z.; Liu, K.J. The interaction of zinc and the blood-brain barrier under physiological and ischemic conditions. Toxicol. Appl. Pharmacol. 2019, 364, 114–119.
- Duck, K.A.; Connor, J.R. Iron uptake and transport across physiological barriers. Biometals 2016, 29, 573–591. Duck. K.A.; Connor, J.R. Iron uptake and transport across physiological barriers. Biometals 2016, 29, 573–591.
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998, 158, 47–52.
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup. G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; Tanzi, R.E.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616.
- Banerjee, P.; Sahoo, A.; Anand, S.; Ganguly, A.; Righi, G.; Bovicelli., P.; Saso, L.; Chakrabarti, S. Multiple mechanisms of iron-induced amyloid beta-peptide accumulation in SHSY5Y cells: Protective action of negletein. Neuromolecular Med. 2014, 16, 787–798. Banerjee, P.; Sahoo.; Anand, S.; Ganguly, A.; Righi, G.; Bovicelli. P.; Saso, L.; Chakrabarti, S. Multiple mechanisms of iron-induced amyloid beta-peptide accumulation in SHSY5Y cells: Protective action of negletein. Neuromolecular Med. 2014, 16, 787–798.
- Suh, S.W.; Jensen, K.B.; Jensen, M.S.; Silva, D.S.; Kesslak, P.J.; Danscher, G.; Frederickson, C.J. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 2000, 852, 274–278. Suh, S.W.; Jensen, K.B.; Jensen, M.S.; Silva, D.S.; Kesslak, P.J.; Danscher, G.; Frederickson, C.J. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 2000, 852, 274–278.
- Zhang, L.H.; Wang, X.; Zheng, Z.H.; Ren, H.; Stoltenberg, M.; Danscher, G.; Huang, L.; Rong, M.; Wang, Z.Y. Altered expression and distribution of zinc transporters in APP/PS1 transgenic mouse brain. Neurobiol. Aging 2010, 31, 74–87. Zhang, L.H.; Wang, X.; Zheng, Z.H.; Ren, H.; Stoltenberg, M.; Danscher, G.; Huang, L.; Rong, M.; Wang, Z.Y. Altered expression and distribution of zinc transporters in APP/PS1 transgenic mouse brain. Neurobiol. Aging 2010, 31, 74–87.
- Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772.
- Hooda, J.; Shah, A.; Zhang, L. Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients 2014, 6, 1080–1102. Hooda, J.; Shah, A.; Zhang, L. Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients 2014, 6, 1080–1102.
- Faux, N.G.; Rembach, A.; Wiley, J.; Ellis, K.A.; Ames, D.; Fowler, C.J.; Martins, R.N.; Pertile, K.K.; Rumble, R.L.; Trounson, B.; et al. An anemia of Alzheimer’s disease. Mol. Psychiatry 2014, 19, 1227–1234. Faux, N.G.; Rembach, A.; Wiley, J.; Ellis, K.A.; Ames, D.; Fowler, C.J.; Martins, R.N.; Pertile, K.K.; Rumble, R.L.; Trounson, B.; et al. An anemia of Alzheimer’s disease. Mol. Psychiatry 2014, 19, 1227–1234.
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Curr. Alzheimer Res. 2016, 13, 174–184. Gozzelino, R. The Pathophysiology of Heme in the Brain. Curr. Alzheimer Res. 2016, 13, 174–184.
- Vanacore, R.; Eskew, J.D.; Sung, L.; Davis, T.; Smith, A. Safe coordinated trafficking of heme and iron with copper maintain cell homeostasis: Modules from the hemopexin system. Biometals 2019, 2, 355–367. Vanacore, R.; Eskew, J.D.; Sung, L.; Davis, T.; Smith, A. Safe coordinated trafficking of heme and iron with copper maintain cell homeostasis: Modules from the hemopexin system. Biometals 2019, 2, 355–367.
- Sankar, S.B.; Donegan, R.K.; Shah, K.J.; Reddi, A.R.; Wood, L.B. Heme and hemoglobin suppress amyloid β-mediated inflammatory activation of mouse astrocytes. J. Biol. Chem. 2018, 293, 11358–11373. Sankar, S.B.; Donegan, R.K.; Shah, K.J.; Reddi, A.R.; Wood, L.B. Heme and hemoglobin suppress amyloid β-mediated inflammatory activation of mouse astrocytes. J. Biol. Chem. 2018, 293, 11358–11373.
- Atamna, H. Heme binding to Amyloid-beta peptide: Mechanistic role in Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 255–266. Atamna, H. Heme binding to Amyloid-beta peptide: Mechanistic role in Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 255–266.
- Atamna, H.; Liu, J.; Ames, B.N. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: Revelance to aging. J. Biol. Chem. 2001, 276, 48410–48416. Atamna, H.; Liu, J.; Ames, B.N. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: Revelance to aging. J. Biol. Chem. 2001, 276, 48410–48416.
- Ghosh, C.; Seal, M.; Mukherjee, S.; Ghosh Dey, S. Alzheimer’s Disease: A Heme-Aβ Perspective. Acc. Chem. Res. 2015, 48, 2556–2564. Ghosh, C.; Seal, M.; Mukherjee, S.; Ghosh Dey, S. Alzheimer’s Disease: A Heme-Aβ Perspective. Acc Chem. Res. 2015, 48, 2556–2564.
- Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L.; Chakrabarti, S. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2021, 2021, 7086512. Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L.; Chakrabarti, S. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2021, 2021, 7086512.
- Frost, G.R.; Jonas, L.A.; Li, Y.M. Friend, Foe or Both? Immune Activity in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 337. Frost, G.R.; Jonas, L.A.; Li, Y.M. Friend, Foe or Both? Immune Activity in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 337.
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172.
- Villa, V.; Thellung, S.; Bajetto, A.; Gatta, E.; Robello, M.; Novelli, F.; Tasso, B.; Tonelli, M.; Florio, T. Novel celecoxib analogues inhibit glial production of prostaglandin E2, nitric oxide, and oxygen radicals reverting the neuroinflammatory responses induced by misfolded prion protein fragment 90-231 or lipopolysaccharide. Pharmacol. Res. 2016, 113, 500–514. Villa, V.; Thellung, S.; Bajetto, A.; Gatta, E.; Robello, M.; Novelli, F.; Tasso, B.; Tonelli, M.; Florio, T. Novel celecoxib analogues inhibit glial production of prostaglandin E2, nitric oxide, and oxygen radicals reverting the neuroinflammatory responses induced by misfolded prion protein fragment 90-231 or lipopolysaccharide. Pharmacol. Res. 2016, 113(Pt A), 500–514.
- Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergström, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J. Neuroinflamm. 2021, 18, 124. Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergström, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J. Neuroinflammation 2021, 18, 124.
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544.
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflammation 2012, 9, 179.
- Xiang, Z.; Haroutunian, V.; Ho, L.; Purohit, D.; Pasinetti, G.M. Microglia activation in the brain as inflammatory biomarker of Alzheimer’s disease neuropathology and clinical dementia. Dis. Markers 2006, 22, 95–102. Xiang, Z.; Haroutunian, V.; Ho, L.; Purohit, D.; Pasinetti, G.M. Microglia activation in the brain as inflammatory biomarker of Alzheimer’s disease neuropathology and clinical dementia. Dis. Markers 2006, 22, 95–102.
- Martínez Leo, E.E.; Segura Campos, M.R. Systemic Oxidative Stress: A key Point in Neurodegeneration—A Review. J. Nutr. Health Aging 2019, 23, 694–699. Martínez Leo, E.E.; Segura Campos, M.R. Systemic Oxidative Stress: A key Point in Neurodegeneration—A Review. J. Nutr. Health Aging. 2019, 23, 694–699.
- Ashour, N.H.; El-Tanbouly, D.M.; El Sayed, N.S.; Khattab, M.M. Roflumilast ameliorates cognitive deficits in a mouse model of amyloidogenesis and tauopathy: Involvement of nitric oxide status, Aβ extrusion transporter ABCB1, and reversal by PKA inhibitor H89. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110366. Ashour, N.H.; El-Tanbouly, D.M.; El Sayed, N.S.; Khattab, M.M. Roflumilast ameliorates cognitive deficits in a mouse model of amyloidogenesis and tauopathy: Involvement of nitric oxide status, Aβ extrusion transporter ABCB1, and reversal by PKA inhibitor H89. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110366.
- Xu, J.J.; Guo, S.; Xue, R.; Xiao, L.; Kou, J.N.; Liu, Y.Q.; Han, J.Y.; Fu, J.J.; Wei, N. Adalimumab ameliorates memory impairments and neuroinflammation in chronic cerebral hypoperfusion rats. Aging 2021, 13, 14001–14014. Xu, J.J.; Guo, S.; Xue, R.; Xiao, L.; Kou, J.N.; Liu, Y.Q.; Han, J.Y.; Fu, J.J.; Wei, N. Adalimumab ameliorates memory impairments and neuroinflammation in chronic cerebral hypoperfusion rats. Aging 2021, 13, 14001–14014.
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener 2016, 11, 23. Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici,M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener 2016, 11, 23.
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572.
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711.
- Chiu, Y.J.; Lin, C.H.; Lee, M.C.; Hsieh-Li, H.M.; Chen, C.M.; Wu, Y.R.; Chang, K.H.; Lee-Chen, G.J. Formulated Chinese medicine Shaoyao Gancao Tang reduces NLRP1 and NLRP3 in Alzheimer’s disease cell and mouse models for neuroprotection and cognitive improvement. Aging 2021, 9, 13. Chiu, Y.J.; Lin, C.H.; Lee, M.C.; Hsieh-Li, H.M.; Chen, C.M.; Wu, Y.R.; Chang, K.H.; Lee-Chen, G.J. Formulated Chinese medicine Shaoyao Gancao Tang reduces NLRP1 and NLRP3 in Alzheimer’s disease cell and mouse models for neuroprotection and cognitive improvement. Aging 2021, 9, 13.
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor-kappa β as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137. Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor-kappa β as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137.
- Ji, Y.; Han, J.; Lee, N.; Yoon, J.H.; Youn, K.; Ha, H.J.; Yoon, E.; Kim, D.H.; Jun, M. Neuroprotective Effects of Baicalein, Wogonin, and Oroxylin A on Amyloid Beta-Induced Toxicity via NF-κB/MAPK Pathway Modulation. Molecules 2020, 25, 5087. Ji, Y.; Han, J.; Lee, N.; Yoon, J.H.; Youn, K.; Ha, H.J.; Yoon, E.; Kim, D.H.; Jun, M. Neuroprotective Effects of Baicalein, Wogonin, and Oroxylin A on Amyloid Beta-Induced Toxicity via NF-κB/MAPK Pathway Modulation. Molecules 2020, 25, 5087.
- Youn, K.; Jun, M. Geraniin Protects PC12 Cells Against Aβ25-35-Mediated Neuronal Damage: Involvement of NF-κB and MAPK Signaling Pathways. J. Med. Food 2020, 23, 928–937.
- Ma, L.Y.; Liu, S.F.; Du, J.H.; Niu, Y.; Hou, P.F.; Shu, Q.; Ma, R.R.; Wu, S.D.; Qu, Q.M.; Lv, Y.L. Chronic ghrelin administration suppresses IKK/NF-κB/BACE1 mediated Aβ production in primary neurons and improves cognitive function via upregulation of PP1 in STZ-diabetic rats. Neurobiol. Learn Mem. 2020, 169, 107155.
More