Leishmaniasis, Chagas disease, and human African trypanosomiasis (HAT), also known as sleeping sickness, are vector borne zoonosis that affect millions of people worldwide and lead to the death of about 100,000 humans per year. Among several molecular targets proposed, trypanothione reductase (TR) is of particular interest for its critical role in controlling the parasite’s redox homeostasis.
1. Introduction
Leishmaniasis, Chagas disease, and human African trypanosomiasis (HAT), also known as sleeping sickness, are vector borne zoonosis that affect millions of people worldwide and lead to the death of about 100,000 humans per year. These diseases are caused by infection with the trypanosomatids Leishmania (L.), Trypanosoma (T.) cruzi, and Trypanosoma brucei, respectively.
Several species of
Leishmania parasites, transmitted by the bite of infected female phlebotomine sandflies, cause three main forms of leishmaniases: visceral (VL), cutaneous (CL), and mucocutaneous (MCL). There are an estimated 700,000 to 100,000,000 new cases of Leishmaniases annually in the world, widely distributed in tropical and subtropical climate zones, which lead to 26,000 to 65,000 deaths
[1].
Chagas disease, also known as American trypanosomiasis, is found mainly in endemic areas of 21 continental Latin American countries, where it affects about 6 to 7 million people. Chagas disease is transmitted to humans by contact with feces or urine of triatomine bugs, known as “kissing bugs”.
Trypanosoma cruzi infection is curable if treatment is initiated soon after infection; if the disease becomes chronic, the patient may develop cardiac, digestive, and/or neurological alterations
[2].
Sleeping sickness is endemic in 36 sub-Saharan African countries, where is transmitted by tsetse flies.
Trypanosoma brucei gambiense accounts for more than 98% of reported cases of the disease. Sustained control efforts have reduced the number of new cases so that in 2009 the number of reported cases dropped below 10,000 for the first time, and in 2018 there were only 977 cases recorded
[3].
These diseases affect some of the poorest countries in the world and are often associated with malnutrition, population migration, poor housing, weak immune systems, such that they are generally recognized as neglected tropical diseases. However, the Mediterranean Basin is included in the affected areas, and climate change will exacerbate the ecological risk of human exposure in regions out of the current range of the disease; therefore, the issue concerns developed countries as well. Moreover, animal infection represents a further socio-economic problem: both domestic and wild animals are a reservoir for human infection, as in the case of endemic canine leishmaniasis in the Mediterranean area; in addition, livestock infection can cause significant economic losses in rural areas, as in the case of Nagana disease in Africa.
The therapeutic arsenal currently available for these diseases includes suramin, pentamidine, melarsoprol, and eflornithine for HAT; benznidazole and nifurtimox for Chagas disease; miltefosine, amphotericin B in liposomal formulation, pentavalent antimonials, and paromomycin for visceral leishmaniasis. Despite the need, these drugs are unsatisfactory because of a number of reasons: they are poorly effective, manifest severe side effects, episodes of resistance are increasingly frequent, and most treatments require prolonged and parenteral administration not suited for therapy in poor countries. Antimonials, for example, have a low therapeutic index and invoke extreme toxicities; therefore, they are administered only if strictly needed, in case of resistance to other treatments. Many different approaches have been attempted to date to develop new trypanocidal drugs, ranging from target based to phenotypic based and repositioning, and some compounds have been moved to clinical trials, but further efforts will be needed for new drugs to hit the market
[4].
Leishmania and
Trypanosoma parasites share many features, including gene conservation, high amino acid identity among proteins, the presence of subcellular structures such as glycosomes and the kinetoplastid, and genome architecture; such conservations may make drug development family-specific, rather than species-specific, i.e., based on the inhibition of a common, conserved target. Many unique metabolic pathways and cellular functions, divergent from other eukaryotes, are attractive target sources for drug discovery
[4].
Oxidative stress plays an essential role in the host immune fight against infection, so that parasite survival mainly depends on the capability to resist this attack
[5]. Infective trypanosomatids lack catalase
[6] and other conventional redox controlling systems
[7][8][7,8], and they base their defense on trypanothione, an unusual variant of glutathione, main actor in maintaining thiols’ homeostasis. Being essential and peculiar, trypanothione is a weakness for these parasites, and all related enzymes are considered interesting candidates for drug development
[9]. Among these, trypanothione reductase (TR), the enzyme directly responsible for keeping trypanothione in the reduced state, has been extensively studied since it fulfills most of the requirements for a good drug target
[10]. Indeed, TR is: (i) essential for parasite survival; (ii) absent in the host, in which TR is replaced by glutathione reductase (GR); (iii) druggable, in that it can be efficiently addressed by inhibitors.
TR has been validated as a target in both
Leishmania and
Trypanosoma as it is not possible to obtain TR-knockout mutants and its downregulation causes strong impairment of infectivity
[11][12][11,12]. It has also been proven that antimonials, among the drugs currently in use to treat leishmaniasis, interfere with the trypanothione metabolism and inhibit TR
[13][14][13,14], reinforcing the idea that targeting this protein is a concrete option for the treatment of these diseases. Moreover, the high sequence homology of TRs from different sources (80–100%) makes it a valuable target for developing a single, broad spectrum drug active against all trypanosomatids
[15].
The main limitation of TR as a drug target lies in its high efficiency/turnover: it was shown that, in order to have a significant effect on parasite redox state and viability, TR activity must be reduced by at least 90%, meaning that only potent inhibitors, with submicromolar IC
50, can be considered very promising lead compounds
[11][12][11,12].
Many efforts have been made in order to find new effective hits through in vitro and in silico screening, in addition to the development of known scaffolds via SAR or structure-based design approaches
[15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33], so that several classes of active compounds have been proposed to date.
2. Relevant Structural Features of TR
Before addressing the binding mode of inhibitors, it is appropriate to describe the main structural features of TR, the mechanism of catalysis, and the recognition of substrates.
The structure of TR is thoroughly characterized, since the crystal structure has been solved for several species, namely
Crithidia fasciculata,
L. infantum,
T. brucei, and
T. cruzi, also in complex with substrates
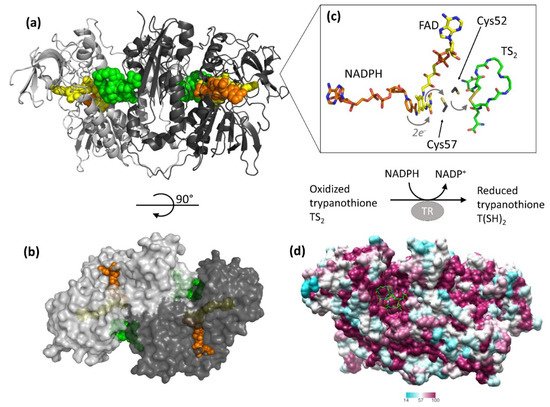
[14][34][35][36][37][14,34,35,36,37]. TR is an obligate homodimer with each of the two individual subunits, related by two-fold symmetry, comprising an FAD-binding domain (residues 1–160 and 289–360), an NADPH-binding domain (residues 161–288), and an interface domain (residues 361–488,
T. brucei numbering).
The protein catalyzes the reduction of the dithiol trypanothione (from TS
2 to T(SH)
2) at the expense of the co-substrate NADPH. NADPH and TS
2 bind different cavities facing opposite sides of the isoallosazine ring of FAD. The TS
2 site, located at the interface between the two subunits, is shaped by residues belonging to both subunits. The reaction mechanism relies on the transfer of two electrons from NADPH to two catalytic cysteines (Cys52 and Cys57), via the FAD cofactor. Once the cysteines are reduced, the oxidized TS
2 binds to the protein, and Cys52, deprotonated by the couple His461′-Glu466′, attacks the disulfide bridge of the substrate, resulting in the formation of a mixed disulfide. Finally, the attack of Cys57 on Cys52 enables the release of the reduced T(SH)
2 (
Figure 1). During catalysis, no major structural changes occur, apart from the strictly necessary displacements of the side chains of the residues involved.
Figure 1. Structure and activity of trypanothione reductase (TR). (a,b) Two views of TR dimer from T. brucei (PDB: 2wow) are shown. NADPH (orange), FAD (yellow), and trypanothione (green) are represented as spheres to highlight the binding sites. (c) The detail shows all entities involved in the electron transfer from NADPH to trypanothione. For clarity, trypanothione is depicted as modeled in TR from T. cruzi (PDB: 1bzl), where a single oxidized conformation is observed in the absence of NADPH. (d) Sequence conservation of TR. The dimer of TR from T. brucei (PDB: 2wow) is colored according to the percentage of amino acid identity with respect to other representative TR sequences (C. fasciculata, T. cruzi, T. congolense, T. brucei, L. braziliensis, L. infantum, L. major). Trypanothione, represented as green sticks, assumes multiple conformations in the wide and highly conserved binding cavity.
The structure is almost identical for all the characterized species, in accordance with the high degree of sequence similarity (
Figure 1, panel D). Indeed, TRs from all Trypanosomatidae share at least 67% of primary sequence, with >82% identity among
Leishmania spp. and >80% among
Trypanosoma spp. Similarity reaches 100% for residues shaping both substrates’ binding sites, with the fact that the mode of binding of ligands is the same for all TRs characterized to date
[34][35][36][34,35,36]. The trypanothione assumes variable conformations in the wide cavity, as an effect of the “dynamics” of its binding. In fact, trypanothione enters as a disulfide but, upon reduction, it is released in an extended conformation. Despite this variability, some interactions emerge to be particularly relevant and specific for binding: Glu18, together with other acidic residues, accounts for the positive charge of the substrate, while the almost hydrophobic patch including Trp21, Tyr110, and Met113 mediates the interaction with the polyamine moiety contained in trypanothione.
This observation suggests that, in the search for new inhibitors, results can be transferred from one TR to the others, and the chance exists to find a common inhibitor active on all TRs that can lead to the development of a broad spectrum trypanocidal drug. This is clearly an ambitious goal because differences in biology and lifestyle of these parasites, although they are closely related genetically, could cause species-specific efficacy, as in the case of eflornithine (DFMO). Indeed, DFMO, a suicide inhibitor of ornithine decarboxylase from any source, is the treatment of choice for advanced stage of the sleeping sickness caused by
T. brucei but is rather ineffective on other infections
[9].
3. Off-Target Evaluation: Comparison with Glutathione Reductase (GR)
Selectivity is a fundamental parameter in the evaluation of a potential pharmacological target. For the development of an antiparasitic drug, it is important to choose a target that has substantial differences compared to the host homolog(s), the so-called off-target(s), in order to promote specific action and minimize side effects.
The trypanothione/TR couple replaces many of the antioxidant and metabolic functions of the glutathione/glutathione reductase (GSH/GR) and thioredoxin/thioredoxin reductase (Trx/TrxT) systems present in the host
[38].
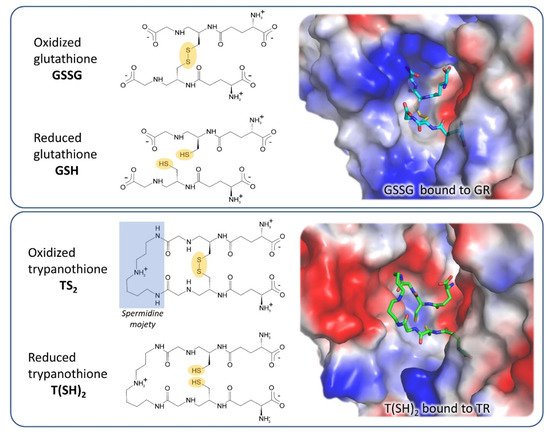
GR is the closest human homolog of TR as they have the same overall fold, with 38% sequence identity, and catalyze the same reaction on very similar substrates. Both GR and TR reduce a disulfide bridge that is intermolecular for GR (GSSG→2 GSH) and intramolecular for TR (TS
2→T(SH)
2). Indeed, trypanothione is an analog of glutathione, comprised of two glutathione molecules linked by amide bonds occurring between the glycyl carboxylate groups of each GSH and the primary amines of the polyamine spermidine.
The most significant differences between the two proteins reflect the differences between their cognate substrates: TS
2 is bulkier than GSSG and positively charged due to the spermidine moiety, while GSH has a net negative charge at physiological pH. As a consequence, the TS
2 binding site in TR is wider and negatively charged with respect to the GSSG binding site in GR (
Figure 2). In particular, selective interactions take place between the spermidine moiety and residues Glu18, Trp21, Ser109, Tyr110, and Met114 that are not conserved in GR and are partially replaced by arginine residues (Arg37, Arg38, and Arg347).
Figure 2. Substrates and active site of glutathione reductase (GR) and TR. The comparison between the electrostatic potential surfaces of GR (upper panel, PDB: 1gra) and TR (lower panel, PDB: 1bzl) highlights the difference in size and charge of substrate binding sites, related to substrates features.
These steric and electrostatic differences account for the selectivity for substrates
[39] and emphasize the potential to generate parasite-specific compounds.
4. Structural Characterization of TR Inhibitors
Structural studies on TR, intensified over the past 10 years, strongly improved the understanding of the molecular basis of ligand binding, allowing to identify hot-spots for interaction with substrates and inhibitors. This knowledge has been exploited in few structure-based design approaches which, in some cases, have led to a significant improvement in the performances of lead molecules
[27][37][40][27,37,40].
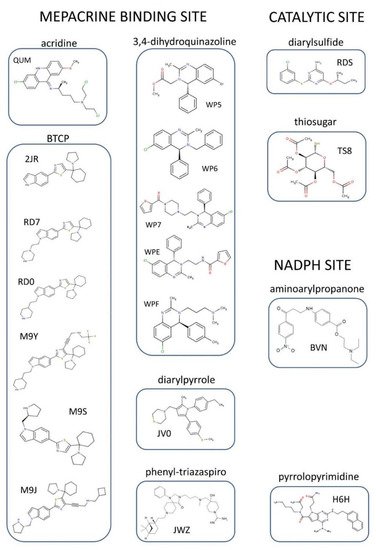
To date, the crystallographic structure of TR in complex with 21 different inhibitors has been defined (see
Table 1 and
Table S1 and Figure 3). These can be grouped into 3 main inhibition modes: (i) competition with trypanothione, due to binding to the wide TS
2 cavity, comprising most of the characterized inhibitors (
Figure 4); (ii) competition with NADPH, due to the binding to NADPH cavity; (iii) redox cysteines inactivation, due to a metal binding to Cys52 and Cys57 in the catalytic site. A fourth inhibition mode has been recently proposed
[41], based on the disassembly of the TR dimer induced by small molecules designed to interfere with protein–protein interaction. However, poor structural information is available for this case.
Figure 3. Organic inhibitors co-crystallized with TR, grouped by binding site and molecular scaffold. Molecules are named by PDB 3-digit ID.
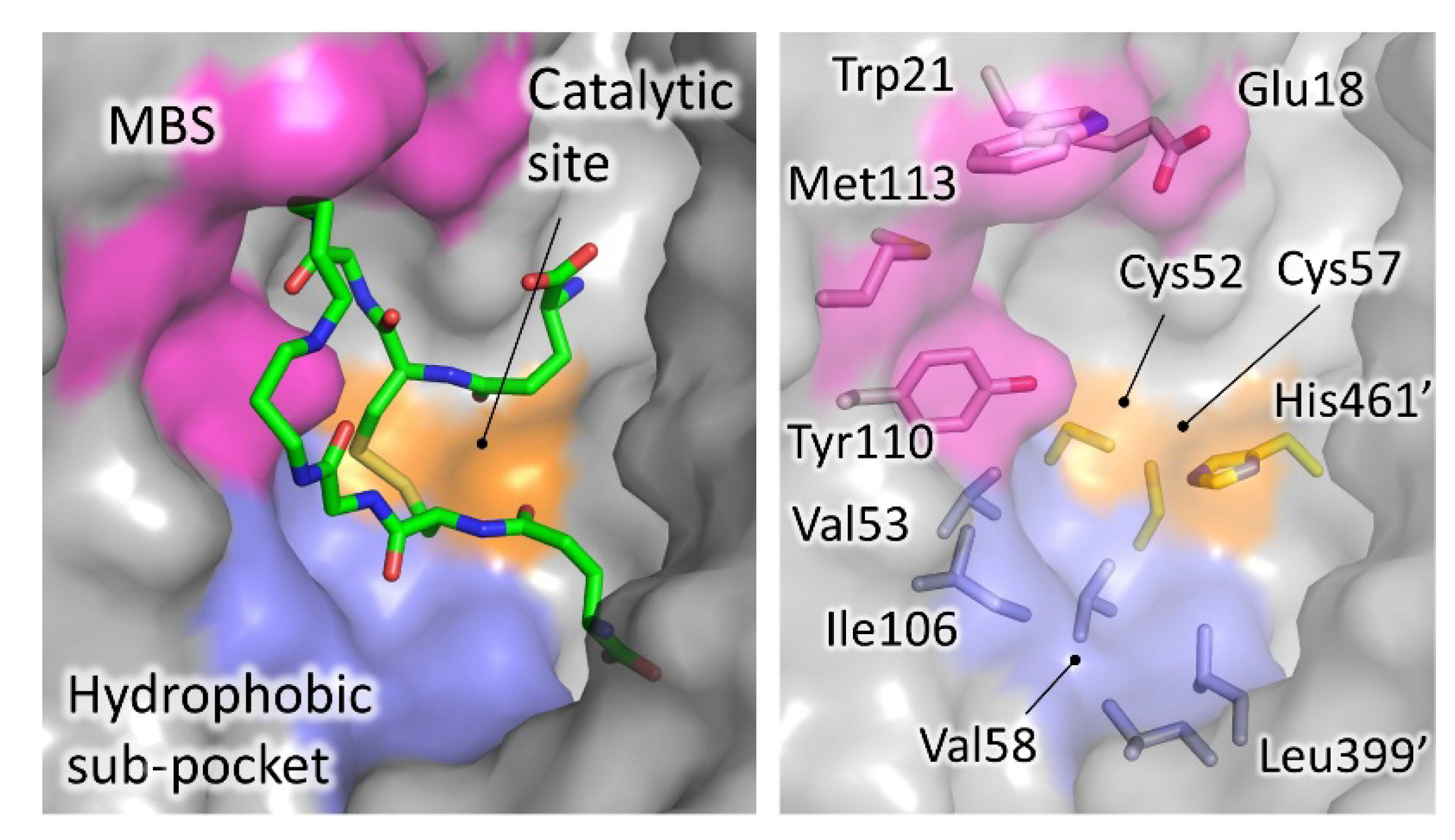
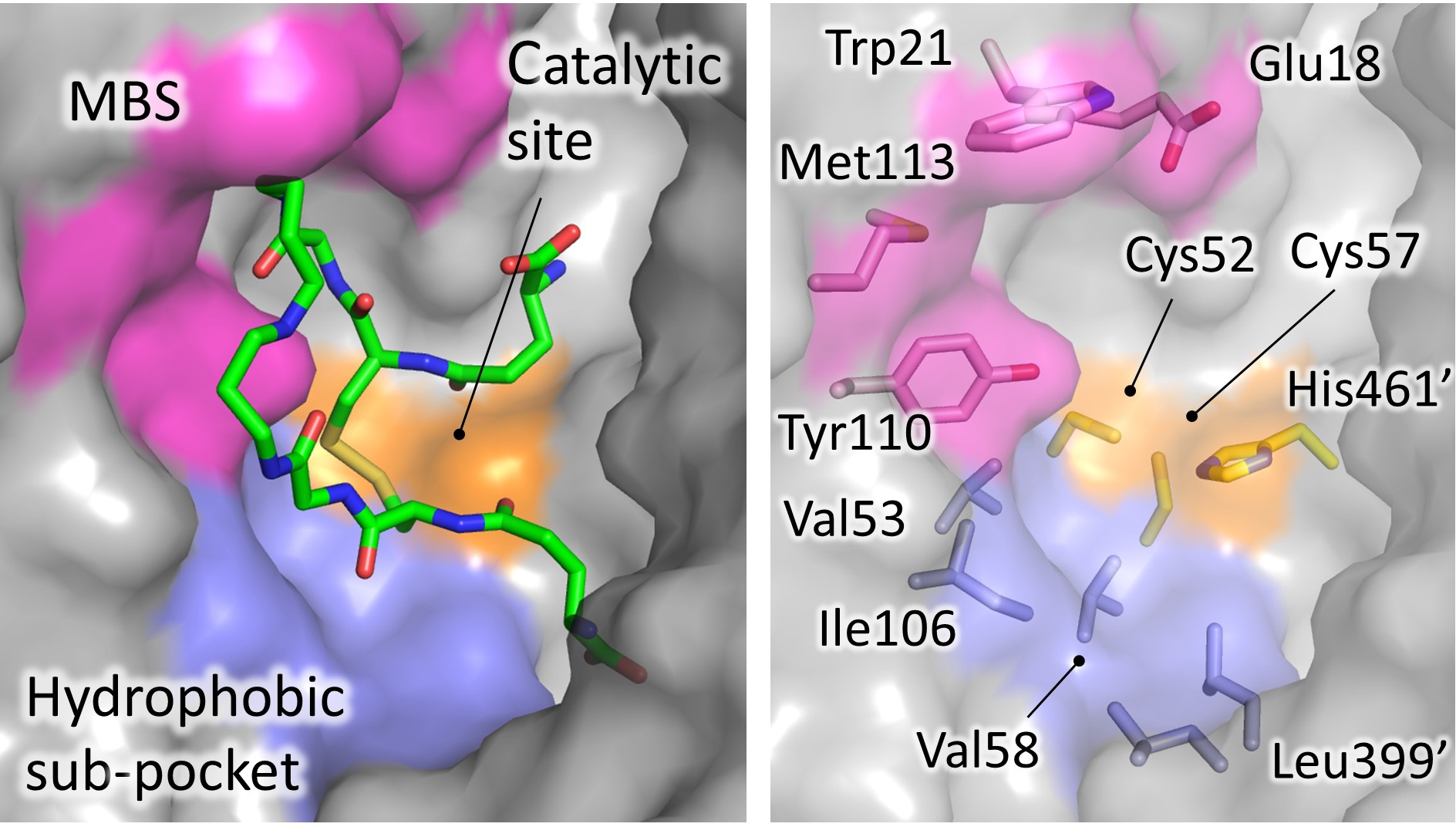 Figure 4. Significant areas for ligand interaction in the Trypanothione binding cavity. Most of the characterized inhibitors bind to in the “mepacrine binding site” (MBS), a hydrophobic patch located at the entrance of the cavity. Fewer ligands bind deeper, in a hydrophobic subpocket closer to the real catalytic site, where the redox cysteines are located and TS2 reduction takes place.
Figure 4. Significant areas for ligand interaction in the Trypanothione binding cavity. Most of the characterized inhibitors bind to in the “mepacrine binding site” (MBS), a hydrophobic patch located at the entrance of the cavity. Fewer ligands bind deeper, in a hydrophobic subpocket closer to the real catalytic site, where the redox cysteines are located and TS2 reduction takes place.
Table 1. All inhibitors co-crystallized with TR.
|
| Site |
|
| Scaffold |
|
| PDB Code |
|
| Source |
|
| Inhibitor PDB ID (Paper ID a) |
|
| Potency b |
|
| Reference |
|
|
| MBS |
|
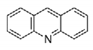
| Acridine |
| 
|
|
| Not available |
|
| Tc |
|
| (Quinacrine or mepacrine) |
|
| Ki: 25 μM |
|
| Jacoby, 1996 |
|
|
| 1gxf |
|
| Tb |
|
| QUM |
| (Quin. mustard) |
|
| Irreversible inhibition |
|
| Saravanamuthu, 2004 |
|
|
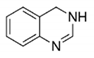
| 3,4-dihydro |
| quinazoline |
| 
|
|
| 2wp5 |
|
| Tb |
|
| WP5 |
| (1a) |
|
| IC50: 6.8 μM |
|
| Patterson, 2011 |
|
|
| 2wp6 |
|
| Tb |
|
| WP6 |
| (6a) |
|
| IC50: 0.93 μM |
|
|
| 2wpc |
|
| Tb |
|
| WP7 |
| (13e) |
|
| IC50: 0.42 μM |
|
|
| 2wpe |
|
| Tb |
|
| WPE |
| (11e) |
|
| IC50: 0.86 μM |
|
|
| 2wpf |
|
| Tb |
|
| WPF |
| (29a) |
|
| IC50: 0.23 μM |
|
|
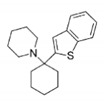
| BTCP |
| 
|
|
| 4nev |
|
| Tb |
|
| 2JR |
| (10a) |
|
| Ki: 12 μM |
| Inh. [%] b: 43 |
|
| Persch, 2014 |
|
|
| 4new |
|
| Tc |
|
| 2JR |
| (10a) |
|
| Ki: 4 μM |
| Inh. [%] b: 79 |
|
|
| 6btl |
|
| Tb |
|
| RD7 |
| (18) |
|
| Ki: 3.8 μM |
| Inh. [%] c: 80 |
|
| De Gasparo, 2018 |
|
|
| 6bu7 |
|
| Tb |
|
| RD0 |
| (19) |
|
| Ki: 6.4 μM |
| Inh. [%] c: 78 |
|
|
| 6oez |
|
| Tb |
|
| M9J |
| ((+)-2) |
|
| Ki: 73 nM |
|
| De Gasparo, 2019 |
|
|
| 6oey |
|
| Tb |
|
| M9S |
| ((+)-4)) |
|
| Ki: 2.1 μM |
|
|
| 6oex |
|
| Tb |
|
| M9Y |
| (5) |
|
| Ki: 1.5 μM |
|
|
| diarylpyrrole |
|
| 4apn (B) |
|
| Li |
|
| JV0 |
| (1) |
|
| Ki: 4.6 μM |
| IC 50: 13.8 μM |
|
| Baiocco, 2013 |
|
|
| phenyl- |
| triazaspiro |
|
| 6br5 |
|
| Tb |
|
| JWZ |
| (1) |
|
| IC50: 5.7 µM |
|
| Turcano, 2020 |
|
|
| Pyrrolopyrimi- dine |
|
| 6i7n |
| (B) |
|
| Li |
|
| H6H |
| (2f) |
|
| IC50: 52.2 µM |
|
| Revuelto, 2019 |
|
|
| Catalytic site |
|
| diaryl sulfide |
|
| 5ebk |
|
| Li |
|
| RDS |
| (RDS 777) |
|
| Ki: 0.25 µM |
|
| Saccoliti, 2017 |
|
|
| Catalytic site/cysteines |
|
| Metal/thiosugar |
|
| 2yau |
|
| Li |
|
| AU-TS8 |
| (auranofin) |
|
| Ki: 0.15 µM |
|
| Ilari, 2012 |
|
|
| Catalytic cysteines |
|
| Metal |
|
| 2w0h |
|
| Li |
|
| SB |
|
| Ki: 1.5 µM |
|
| Baiocco, 2009 |
|
|
| 2x50 |
|
| Li |
|
| AG |
|
| Ki (Ag1): 500 nM |
| K i (Ag0): 50 nM |
|
| Baiocco, 2010 |
|
|
| NADPH-cavity |
|
| 3-amino-1-arylpropan-1-one |
|
| 6er5 |
|
| Li |
|
| BVN |
| (3) |
|
| IC50: 12.4 µM |
|
| Turcano, 2018 |
|
a identification code of the inhibitor as reported in the original paper. b Ki, IC50 and/or percentage of inhibition are reported when available in literature. c Percent inhibition by 40 mM inhibitor in the presence of 40 mM dithiol trypanothione (TS2).