Elastin is expressed in the lung by several types of cells, including pleural mesothelial cells, airway and blood vessel smooth muscle cells, endothelial cells, and interstitial fibroblasts. Due to this, a system of elastic fibers is formed, which ensures an equal transfer of the applied force to all parts of the lung
[23][91]. Numerous studies show that the formation of alveoli is closely related to the formation of elastin fibers in the respiratory region of the lung. During septation, the formation of elastin fibers reaches its peak. Three phases of tropoelastin expression have been observed in the developing lung, with each phase characterized by a distinct expression pattern
[24][50]. In the first phase, on E15.5, the expression of tropoelastin mRNA was confined to mesenchymal cells lining the proximal tubules of the endoderm. During the second phase of expression, at the saccular stage on E18.5, almost all lung mesenchyme was tropoelastin-positive. It was also expressed in the developing vascular walls. In the third phase, on P7–P14, tropoelastin expression was limited to a population of mesenchymal cells located on the tips of the growing alveolar septa (and to developing cells of the vascular and bronchial walls). Mesenchymal tropoelastin-positive cells detected in the third phase of postnatal expression were specifically and completely absent in the lung of PDGF-A
−/− mice, while tropoelastin expression in bronchial and blood vessel walls was unaffected. The impairment of postnatal alveologenesis in PDGF-A
−/− mice was suggested to be due to a prenatal block in the distal distribution of PDGFRα
+ cells
[24][50]. In the absence of a PDGF-A signal, PDGFRα
+ cells are unable to multiply and spread. Thus, the depletion of myofibroblasts in PDGF-A-null mice results in a lack of elastic fibers in the alveolar walls and incomplete alveolarization
[25][24][27,50]. Similar effects were observed in Hoxa5
−/− murine lung, in which the subset of the mesenchymal progenitors of alveolar αSMA
+ myofibroblasts expressing Pdgfrα failed to spread into the distal fetal lung
[26][152]. They were trapped in the parenchyma surrounding future alveoli, where they produced abnormal elastic fibers. Elastic fibers were disorganized and aberrantly distributed within the pulmonary tissue on P0 and P5, and alveologenesis was impaired. From P15 onwards, the fibers appeared more abundant, disorganized, and fragmented. Northern blot analyses of tropoelastin expression failed to reveal differences in tropoelastin transcript levels between wild-type and Hoxa5
−/− lungs. Later, it was shown that Hox5 genes are required in elastin network formation by regulating the adherence of the alveolar fibroblasts to fibronectin, which is disrupted because of the loss of the integrin heterodimer Itga5b1 in mutant fibroblasts
[27][153]. In Hoxa5 mutants, the activated macrophages were abundantly found in the lung as early as birth, and most of them expressed matrix metalloproteinase 12 (MMP12). This recruitment coincided with the abnormal elastin deposition by the mispositioned alveolar myofibroblasts, supporting the notion that elastin fragments are chemotactic for monocytes
[28][154]. Furthermore, recent studies showed that the absence of Pdgfra in Gli1
+ SCMFs disrupted the expression of elastogenic genes, including members of the LOX, fibronectin, and fibulin families, while the expression of the EGF family members was increased, and Tgfb1 was suppressed in the lung fibroblasts of the studied mice. Similar results were found in lung samples from a person with BPD. Blocking PDGF-A signaling in vitro had the same effect. In addition, the effect was reversible by inhibiting EGF or activating TGF-beta signaling. These data demonstrate the role of PDGFA/PDGFRα in the control of elastogenic gene expression through the secondary level of signaling networks consisting of EGF and TGF-beta
[29][80]. Finally, the growth factor FGF-18 was also shown to affect myofibroblast differentiation by stimulating the expression of tropoelastin, LOX, and fibulins 1 and 5 during secondary septation
[30][51].
Some methods that suppress alveolarization, such as prolonged hyperoxia (BPD modeling), also suppress septation and elastin expression in the alveolar wall
[31][32][33][34][155,156,157,158]. Elastin fiber assembly impairment in newborn rats due to copper deficiency
[35][159] or the administration of beta-Aminopropionitrile (beta APN)
[36][160], which interferes with the synthesis of elastin and collagen by inhibiting LOX, results in the impairment of alveolar septa formation and the simplification of the alveoli by the type of emphysema. In contrast to PDGF-A
−/− mice, in which the absence of alveolar elastin synthesis and the abnormal structure of the alveoli are detected after P4, the inactivation of the elastin gene in Eln
−/− mice leads to a delay in the perinatal development of the terminal airway branches. At birth, these mice have enlarged cavities in the lung that resemble emphysema
[37][161]. These differences indicate that, in addition to its role in the formation of the structure of alveoli, elastin is important for the terminal branching of the airways in lung development. This also supports the idea that elastin is expressed by airway smooth muscle cells, which enwrap the branching tips and the stalk of the primordial airways and physically support branching morphogenesis
[38][39][162,163].
In the developing fetal lung, the interplay between the epithelium and the mesenchyme surrounding it is a driving force that guides the formation of the proximal–distal pattern. Respiratory mesoderm originates from the cardio-pulmonary precursors expressing Wnt2, Gli1, and Isl1 and generates vascular and airway smooth muscle cells, proximal vascular endothelium, and pericyte-like cells
[40][11]. Tbx4 is expressed in the population of the mesenchymal cells that give rise to the entire stroma of the postnatal lung. TBX4-positive mesenchymal precursor cells proliferate and differentiate into different cell types, simultaneously cooperating with the developing lung epithelium
[41][42][12,13]. In the regions adjacent to the endoderm of the distal tip, mesodermal cells express FGF10, which stimulates branching morphogenesis
[43][44][45][46][14,15,16,17]. In the fetal and postnatal lung, FGF10
+ cells represent the pool of mesenchymal progenitor cells
[47][18]. The endoderm of the distal tip expresses SOX9 and ID2. Endodermal cells respond to WNT and the chemoattracting and morphogenic action of mesodermal FGF10 (for the review, see
[48][19]). The Shh and Ptch1 expression patterns are the reverse of the expression patterns of Fgf10 and Fgfr2b in that the Shh ligand is expressed in the epithelium, which is of endodermal origin, while the Ptch1 receptor is expressed in the mesenchyme
[49][20]. The formation of a vast branching network of airways is partly explained by a signaling mechanism based on a ligand–receptor interplay between Fgf10 and Shh, which directs the outgrowth of the lung bud via a ligand–receptor-based Turing mechanism, and is additionally determined by a geometry effect of the lung
[50][21]. The lung mesenchyme possesses positional information and anatomical specificity: proximal and distal mesenchyme can induce the corresponding proximal and distal differentiation of the lung epithelium
[7][8][7,8]. In 1994, Shannon was already able to show the induction of alveolar type II cell differentiation in fetal tracheal epithelium by the transplanted distal lung mesenchyme
[7].
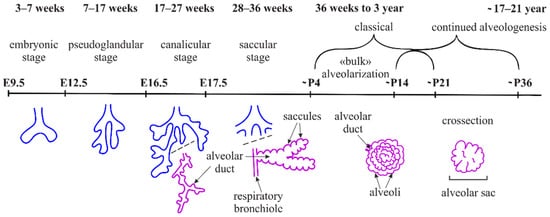
During the pseudoglandular stage, the recurrent processes of outgrowth and branching of the lung endoderm (branching morphogenesis) are initialized, by which all the future respiratory airways (bronchi and bronchioli) are formed (
Figure 1). It is interesting to note that the specification of alveolocyte clones occurs earlier than it was supposed before; more specifically, it runs in parallel to the processes of tissue patterning, including the formation of the axial endoderm pattern and branching morphogenesis
[51][22]. At the end of branching morphogenesis on E15.5, precursors of the alveolar type 1 and 2 cells (AT1 and AT2) are detected in the bud tips
[5]. During the canalicular phase, the differentiation of the epithelium can be detected (via alveolocyte marker staining), and the junctions of bronchiolar ducts, which are the extensions of the conducting airways, are formed. Further in development, the enlargements (saccules) are formed along the branching tree, thus marking the saccular phase (
Figure 1). Separations between initial alveolar sacs are known as primary septa. Several authors also designate this phase as initial alveologenesis. Primary septa are not effective in the formation of the air–blood barrier and full breathing. Thus, the survival of an individual depends on the timely and accurate formation of the secondary septa. Mice are born in the saccular phase of lung development. The formation of their secondary septa and alveoli occurs after birth. In humans, alveologenesis begins in utero on the 36th week of gestation (
Figure 1), and classical alveologenesis continues for 2–3 years after birth, when the number of alveoli increases. Afterwards, from 3 until 21 years, the size of alveoli increases
[52][23].
4. Postnatal Alveologenesis
The formation of the alveoli (alveologenesis) is the final stage of lung development and is subdivided into two phases (
Figure 1). The first phase begins on the 3rd–4th day of postnatal development (P3–P4), and is characterized by the formation of the secondary septa, which represent the invaginations of the saccular walls rich in ECM and separate and expand the alveolar surface area
[53][24]. Around P3, as the breathing mode is established, a subpopulation of the secondary crest myofibroblasts (SCMFs, also known as alveolar myofibroblasts) with increased traction force appears in the lung mesenchyme
[5]. SCMFs participate in secondary septa formation and actively accept different signals from AT1 cells. Contractile activity of myofibroblasts is supposed to physically form alveoli
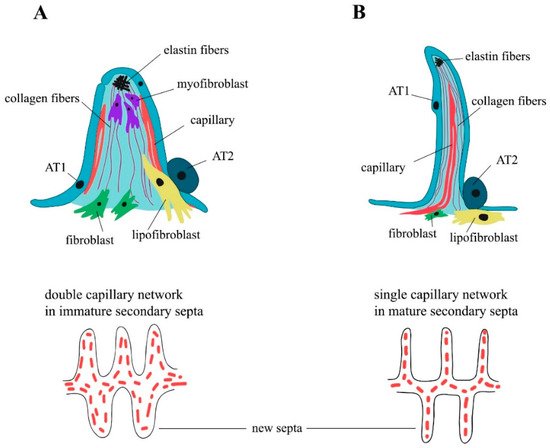
[54][25]. SCMF precursors are the subpopulation of platelet-derived growth factor receptor α (PDGFRα) positive cells that later in embryonic development become α-smooth muscle actin (αSMA) positive and are found on the tips of the alveolar septa in close proximity to deposits of elastin (
Figure 2A).
Figure 2. Alveolar secondary septa. Classical alveologenesis. Immature, thick secondary septa. Double capillary network (
A). Continued alveologenesis. Mature, thin secondary septa. Single capillary network (
B). Redrawn from
[52][55][23,26].
PDGF-A and its sole receptor PDGFRα constitute an axis of cross-communication between pulmonary endodermal and mesodermal cells
[25][27]. Endodermal cells express the ligand
[25][27], and PDGFRα is ubiquitously expressed throughout the lung mesoderm
[56][57][28,29]. Earlier, PDGF-A signaling over PDGFRα was shown to be necessary for lung growth and alveoli formation, but not for branching morphogenesis
[58][30]. According to a widespread model of septa formation, PDGF-A secreted by the epithelium is extruded into the lumen of alveoli by the exerting force of myofibroblasts and developing capillary. Then, the septa are thinned through the processes of apoptosis and capillary maturation
[59][60][31,32]. Impaired Notch signaling in mice, especially Notch2, results in the abnormal enlargement of the alveoli. During neonatal life, Notch2 is activated in AT2 cells to induce PDGF-A expression, triggering the paracrine activation of PDGFRα signaling in the SCMF precursors required for alveologenesis
[61][33]. The first phase of alveologenesis, from around P5 to P14, when SCMFs are present, is called classical alveologenesis (
Figure 2A). During the second phase of alveologenesis (continued alveologenesis), from around P14 to P36, formed immature septa start thinning, the double capillary septal network is modified to a single one, thus forming mature, thin septa in an adult lung (
Figure 2B)
[60][32]. After the active phase of alveologenesis is completed, more than 20% of interstitial fibroblasts are subjected to apoptosis, a process that is crucial for the normal development of the lung
[62][63][34,35]. Nevertheless, taking into account the cell heterogeneity of the mesenchymal compartment, the question of which subpopulation of fibroblasts is eliminated thus becomes controversial
[62][63][64][34,35,36]. There is an opinion that the thinning of the alveolar walls during the second phase of alveologenesis is caused by the depletion of lung myofibroblasts after around P15 through apoptosis and phagocytosis
[65][37]. The flow cytometry evaluation of lipid-filled interstitial fibroblasts stained with a lipophilic marker showed that this exact subpopulation is eliminated via apoptosis after alveolarization
[66][38]. Results of several studies have suggested the temporal expression of αSMA by some subpopulations of alveolar mesenchymal cells that play the role of SCMFs and are not eliminated afterwards
[67][68][39,40]. Multiple data have demonstrated that myofibroblasts are crucial for secondary septa formation, but the mechanisms of αSMA
+ SCMFs depletion after the first phase, and the question of which cells and mechanisms participate in the second phase of alveologenesis after the elimination of αSMA
+ SCMFs by P15, are the subject of future investigations.
Alveolar density reaches its peak on P39 and stays unchanged after 9 months. The generation of alveoli is fast between P5 and P14, but slows down afterwards
[69][41]. As mentioned before, immature secondary septa have a double capillary network and are quite ineffective in the required gas exchange (
Figure 2A). During the maturation of the blood microvessels, the septa start thinning and two capillary layers merge and thus form a more effective single capillary network (
Figure 2B) (for the review, see
[52][23]). The single capillary network exchanges oxygen and carbon dioxide with two adjacent alveoli, which adds to the efficacy of the gas exchange. On P4–P21, new secondary septa are formed from immature secondary septa emerging at first from the walls of saccules, and further in development from the walls of immature secondary septa with the double capillary network. On days P14–P36, new septa are lifted off mature septa containing single capillary networks (
Figure 2). At the same time, the local duplication of the capillary network due to angiogenesis is found at the base of the newly formed septa in the second phase of alveologenesis. The alveolarization and maturation of microvessels are the processes that run in parallel
[70][42]. Lung development continues into young adulthood: for mice, data are known up to P39
[69][41], for rats, up to P60
[71][43]. Alveoli are, in fact, able to form at any time and anywhere inside the lung parenchyma
[52][23]. Thus, in the alveolar septum, the air–blood barrier has thick parts that contain the nuclei of AT1 cells and fibroblasts and the fiber network of ECM, which provides the regenerative capacity and mechanical stability of the septum; it also has thin parts where the alveolar epithelium and the capillary endothelium share one common basal lamina that minimize the thickness of the diffusion barrier. In the human lung, about half of the entire air–blood barrier is thin
[72][73][44,45].