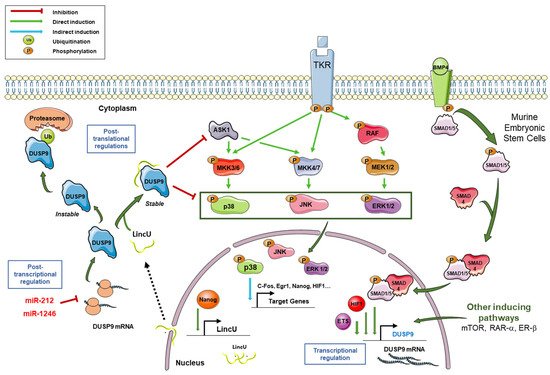
Dual-specificity phosphatase 9 (DUSP9) belongs to the threonine/tyrosine dual-specific phosphatase family and was first described in 1997, is known to dephosphorylate ERK1/2, p38, JNK and ASK1, and thereby to control various MAPK pathway cascades. As a consequence, DUSP9 plays a major role in human pathologies and more specifically in cardiac dysfunction, liver metabolic syndromes, diabetes, obesity and cancer including drug response and cell stemness.
- mitogen-activated protein kinase
- dual-specificity phosphatase
- MAP kinase phosphatase
1. Introduction
The mitogen-activated protein kinase (MAPK) signaling pathways are crucial in cell function and homeostasis. MAPKs regulate pathophysiological processes by controlling signal translation and cellular response such as survival, proliferation, differentiation and migration [1,2,3][1][2][3]. They are activated by a double phosphorylation process on tyrosine and threonine residues in a conserved Thr-X-Tyr motif (X being any amino acid) [4,5][4][5]. Activation of MAPK pathways triggers multiple intracellular signaling cascades. Each cascade is initiated by a specific signal and leads to the activation of a particular MAPK [6]. Once activated, MAPK can phosphorylate various cytoplasmic and/or nuclear substrates and induce changes in the function of target proteins and gene expression [6]. Spatial localization of MAPKs also determines the target substrates and subsequent cellular effects [7].
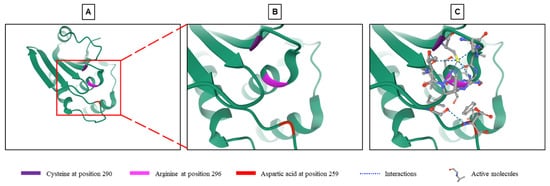
Phosphatases reverse phosphorylation and return MAPKs to an inactive state. The dual specificity phosphatase (DUSP) family belongs to the 199 phosphatases encoded in the human genome. This family is composed of 61 phosphatases capable of downregulating MAPKs by dephosphorylating both tyrosine and serine/threonine residues in a single substrate [12,15][8][9]. The phosphorylation of proteins is a reversible process. This prevents the abnormal activation of the signal and fine-tunes its activity and downstream effects [3,12][3][8]. The balance between phosphorylation and dephosphorylation controls the expression, function, activity and localization of many proteins [12,16][8][10]. Dephosphorylation by DUSPs regulates the duration, intensity and spatiotemporal profile of the MAPK signaling cascade [17][11]. This dephosphorylation takes place thanks to the highly conserved phosphatase site which contains arginine, cysteine and aspartic acid [3,12,18][3][8][12]. In addition to the active site common to all DUSPs, some DUSPs contain a MAP kinase-binding motif (MKB), also called a kinase-interacting motif (KIM), which interacts with the common docking domain of MAPKs to allow the interaction between the enzyme and the substrate [3,12,18,19][3][8][12][13]. Ten DUSPs containing the KIM domain are classified as typical DUSPs or MAP kinase phosphatases (MKPs) (Table 1), while those which do not have this domain (16 phosphatases in total) are called atypical DUSPs [3,5,20][3][5][14]. However, there are a few exceptions. DUSP2, DUSP5 and DUSP8 are typical DUSPs and contain the KIM domain but they are not called MKPs. On the other hand, DUSP14 and DUSP26, which are atypical DUSPs and do not contain a KIM domain, are called MKP6 and MKP8, respectively (Table 1) [3,20][3][14]. Typical DUSPs are the best characterized within the DUSP family [20][14] and this comprises the typical DUSP named DUSP9 or MKP4, which was first described in 1997 by Muda and collaborators [18][12]. This 42-kDa protein dephosphorylates several substrates including JNK, p38, the MAPKKK apoptosis signal-regulating kinase 1 (ASK1) and ERK1/2 with a high specificity for ERK kinases [3,18][3][12].
| Classification | Gene Symbol | Synonyms | Chromosomal Localization | Cell Localization | MAPK Substrates (Others) | Inducible by MAPKs | Main Functions in Physiological and Pathophysiological States |
|---|---|---|---|---|---|---|---|
| Typical MKPs | DUSP1 | MKP1 | 5 | Nuclear | JNK, p38 > ERK | ERK, p38 | Involved in infectious diseases, pulmonary diseases, inflammatory disorders, atherosclerosis, tumorigenesis and tumor progression [15]. |
| DUSP2 | PAC1 | 2 | Nuclear | ERK, JNK, p38 | ERK, JNK | Involved in immune and inflammatory responses, cancer, CLN3 disease and endometriosis [16]. | |
| DUSP4 | MKP2 | 8 | Nuclear | ERK, JNK > p38 | ERK | Involved in inflammatory cytokine secretion, susceptibility to sepsis shock, and resistance to Leishmania mexicana infection [17][18]. | |
| DUSP5 | hVH3 | 10 | Nuclear | ERK | ERK | Plays an anti-inflammatory role and has tumor suppressive functions in several types of cancer [19]. | |
| DUSP6 | MKP3 | 12 | Cytoplasmic | ERK | ERK | Plays a role in carcinogenesis in several cancers as an oncogene or a tumor suppressor [20]. | |
| DUSP7 | MKPX | 3 | Cytoplasmic | ERK, JNK, p38 | N/D | Involved in some cancers [21]. | |
| DUSP8 | hVH5 | 11 | Dually-located | ERK, JNK, p38 | N/D | Plays a role in the central nervous system, circulatory system, urinary system, immune system, genetic diseases and cancers [22]. | |
| DUSP9 | MKP4 | X | Cytoplasmic | ERK >> p38, JNK | N/D | Involved in development of cardiac dystrophy, metabolic diseases and cancers [23][24][25][26][27]. | |
| (MAP3K5/ASK1) | |||||||
| DUSP10 | MKP5 | 1 | Dually-located | JNK, p38 >> ERK | N/D | Involved in immune response, anti-inflammatory response and some cancers [28]. | |
| DUSP16 | MKP7 | 12 | Dually-located | JNK | N/D | Involved in non-alcoholic steatohepatitis and some cancers [29]. | |
| Atypical MKPs | DUSP14 | MKP6 | 17 | Dually-located | ERK, JNK, p38 | N/D | Involved in immune response, bone diseases and cancers [30]. |
| DUSP26 | MKP8 | 8 | Nuclear | p38 | N/D | Regulates neuronal cell proliferation and acts as an oncogene or a tumor suppressor depending on the cellular context [31]. |
2. General Characteristics of DUSP9 and Mechanisms of Regulation
References
- Keshet, Y.; Seger, R. The MAP Kinase Signaling Cascades: A System of Hundreds of Components Regulates a Diverse Array of Physiological Functions. In MAP Kinase Signaling Protocols; Seger, R., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 661, pp. 3–38. ISBN 978-1-60761-794-5.
- Jiang, L.; Wang, Y.; Liu, G.; Liu, H.; Zhu, F.; Ji, H.; Li, B. C-Phycocyanin Exerts Anti-Cancer Effects via the MAPK Signaling Pathway in MDA-MB-231 Cells. Cancer Cell Int. 2018, 18, 12.
- Chen, H.-F.; Chuang, H.-C.; Tan, T.-H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668.
- Cuenda, A.; Rousseau, S. P38 MAP-Kinases Pathway Regulation, Function and Role in Human Diseases. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2007, 1773, 1358–1375.
- Gaestel, M. MAPK-Activated Protein Kinases (MKs): Novel Insights and Challenges. Front. Cell Dev. Biol. 2016, 3, 88.
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254.
- Burotto, M.; Chiou, V.L.; Lee, J.-M.; Kohn, E.C. The MAPK Pathway across Different Malignancies: A New Perspective: Tissue-Specific MAPK Signaling. Cancer 2014, 120, 3446–3456.
- Nayak, J.; Gastonguay, A.J.; Talipov, M.R.; Vakeel, P.; Span, E.A.; Kalous, K.S.; Kutty, R.G.; Jensen, D.R.; Pokkuluri, P.R.; Sem, D.S.; et al. Protein Expression, Characterization and Activity Comparisons of Wild Type and Mutant DUSP5 Proteins. BMC Biochem. 2014, 15, 27.
- Shen, Z.; Zhang, C.; Qu, L.; Lu, C.; Xiao, M.; Ni, R.; Liu, J. MKP-4 Suppresses Hepatocarcinogenesis by Targeting ERK1/2 Pathway. Cancer Cell Int. 2019, 19, 61.
- Huang, C.-Y.; Tan, T.-H. DUSPs, to MAP Kinases and Beyond. Cell Biosci. 2012, 2, 24.
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and P38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202.
- Muda, M.; Boschert, U.; Smith, A.; Antonsson, B.; Gillieron, C.; Chabert, C.; Camps, M.; Martinou, I.; Ashworth, A.; Arkinstall, S. Molecular Cloning and Functional Characterization of a Novel Mitogen-Activated Protein Kinase Phosphatase, MKP-4. J. Biol. Chem. 1997, 272, 5141–5151.
- Camps, M.; Nichols, A.; Gillieron, C.; Antonsson, B.; Muda, M.; Chabert, C.; Boschert, U.; Arkinstall, S. Catalytic Activation of the Phosphatase MKP-3 by ERK2 Mitogen-Activated Protein Kinase. Science 1998, 280, 1262–1265.
- Buffet, C. Anomalies Moléculaires de la Voie MAPK et Cancer Papillaire de la Thyroïde: Étude de Deux Phosphatases Spécifiques de ERK, DUSP5 et DUSP6. Ph.D. Thesis, Université René Descartes, Paris, France, 2015.
- Shen, J.; Zhang, Y.; Yu, H.; Shen, B.; Liang, Y.; Jin, R.; Liu, X.; Shi, L.; Cai, X. Role of DUSP1/MKP1 in Tumorigenesis, Tumor Progression and Therapy. Cancer Med. 2016, 5, 2061–2068.
- Wei, W.; Jiao, Y.; Postlethwaite, A.; Stuart, J.M.; Wang, Y.; Sun, D.; Gu, W. Dual-Specificity Phosphatases 2: Surprising Positive Effect at the Molecular Level and a Potential Biomarker of Diseases. Genes Immun. 2013, 14, 1–6.
- Hsiao, W.-Y.; Lin, Y.-C.; Liao, F.-H.; Chan, Y.-C.; Huang, C.-Y. Dual-Specificity Phosphatase 4 Regulates STAT5 Protein Stability and Helper T Cell Polarization*. PLoS ONE 2015, 10, e0145880.
- Menyhart, O.; Budczies, J.; Munkácsy, G.; Esteva, F.J.; Szabó, A.; Miquel, T.P.; Győrffy, B. DUSP4 Is Associated with Increased Resistance against Anti-HER2 Therapy in Breast Cancer. Oncotarget 2017, 8, 77207–77218.
- Seo, H.; Cho, Y.-C.; Ju, A.; Lee, S.; Park, B.C.; Park, S.G.; Kim, J.-H.; Kim, K.; Cho, S. Dual-Specificity Phosphatase 5 Acts as an Anti-Inflammatory Regulator by Inhibiting the ERK and NF-ΚB Signaling Pathways. Sci. Rep. 2017, 7, 17348.
- Muhammad, K.A.; Nur, A.A.; Nurul, H.S.; Narazah, M.Y.; Siti, R.A.R. Dual-Specificity Phosphatase 6 (DUSP6): A Review of Its Molecular Characteristics and Clinical Relevance in Cancer. Cancer Biol. Med. 2018, 15, 14.
- Luan, T.; Zhang, X.; Wang, S.; Song, Y.; Zhou, S.; Lin, J.; An, W.; Yuan, W.; Yang, Y.; Cai, H.; et al. Long Non-Coding RNA MIAT Promotes Breast Cancer Progression and Functions as CeRNA to Regulate DUSP7 Expression by Sponging MiR-155-5p. Oncotarget 2017, 8, 76153–76164.
- Ding, T.; Zhou, Y.; Long, R.; Chen, C.; Zhao, J.; Cui, P.; Guo, M.; Liang, G.; Xu, L. DUSP8 Phosphatase: Structure, Functions, Expression Regulation and the Role in Human Diseases. Cell Biosci. 2019, 9, 70.
- Jiang, L.; Ren, L.; Guo, X.; Zhao, J.; Zhang, H.; Chen, S.; Le, S.; Liu, H.; Ye, P.; Chen, M.; et al. Dual-Specificity Phosphatase 9 Protects against Cardiac Hypertrophy by Targeting ASK1. Int. J. Biol. Sci. 2021, 17, 2193–2204.
- Li, Z.; Fei, T.; Zhang, J.; Zhu, G.; Wang, L.; Lu, D.; Chi, X.; Teng, Y.; Hou, N.; Yang, X.; et al. BMP4 Signaling Acts via Dual-Specificity Phosphatase 9 to Control ERK Activity in Mouse Embryonic Stem Cells. Cell Stem Cell 2012, 10, 171–182.
- Wei, Q.; Pu, X.; Zhang, L.; Xu, Y.; Duan, M.; Wang, Y. Expression of Dual-Specificity Phosphatase 9 in Placenta and Its Relationship with Gestational Diabetes Mellitus. J. Diabetes Res. 2019, 2019, 1–7.
- Wu, S.; Wang, Y.; Sun, L.; Zhang, Z.; Jiang, Z.; Qin, Z.; Han, H.; Liu, Z.; Li, X.; Tang, A.; et al. Decreased Expression of Dual-Specificity Phosphatase 9 Is Associated with Poor Prognosis in Clear Cell Renal Cell Carcinoma. BMC Cancer 2011, 11, 413.
- Ye, P.; Xiang, M.; Liao, H.; Liu, J.; Luo, H.; Wang, Y.; Huang, L.; Chen, M.; Xia, J. Dual-Specificity Phosphatase 9 Protects Against Nonalcoholic Fatty Liver Disease in Mice through ASK1 Suppression: Steatohepatitis/Metabolic Liver Disease. Hepatology 2019, 69, 76–93.
- Jimenez, T.; Barrios, A.; Tucker, A.; Collazo, J.; Arias, N.; Fazel, S.; Halim, M.; Huynh, T.; Singh, R.; Pervin, S. DUSP9-Mediated Reduction of PERK1/2 Supports Cancer Stem Cell-like Traits and Promotes Triple Negative Breast Cancer. Am. J. Cancer Res. 2020, 10, 3487–3506.
- Wu, Y.-K.; Hu, L.-F.; Lou, D.-S.; Wang, B.-C.; Tan, J. Targeting DUSP16/TAK1 Signaling Alleviates Hepatic Dyslipidemia and Inflammation in High Fat Diet (HFD)-Challenged Mice through Suppressing JNK MAPK. Biochem. Biophys. Res. Commun. 2020, 524, 142–149.
- Yang, C.-Y.; Li, J.-P.; Chiu, L.-L.; Lan, J.-L.; Chen, D.-Y.; Chuang, H.-C.; Huang, C.-Y.; Tan, T.-H. Dual-Specificity Phosphatase 14 (DUSP14/MKP6) Negatively Regulates TCR Signaling by Inhibiting TAB1 Activation. J. Immunol. 2014, 192, 1547–1557.
- Jung, S.; Nah, J.; Han, J.; Choi, S.-G.; Kim, H.; Park, J.; Pyo, H.-K.; Jung, Y.-K. Dual-Specificity Phosphatase 26 (DUSP26) Stimulates Aβ42 Generation by Promoting Amyloid Precursor Protein Axonal Transport during Hypoxia. J. Neurochem. 2016, 137, 770–781.
- Low, H.B.; Zhang, Y. Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 2016, 16, 85–98.
- Seternes, O.-M.; Kidger, A.M.; Keyse, S.M. Dual-Specificity MAP Kinase Phosphatases in Health and Disease. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2019, 1866, 124–143.
- Keyse, S.M. Dual-Specificity MAP Kinase Phosphatases (MKPs) and Cancer. Cancer Metastasis Rev. 2008, 27, 253–261.
- Lang, R.; Hammer, M.; Mages, J. DUSP Meet Immunology: Dual Specificity MAPK Phosphatases in Control of the Inflammatory Response. J. Immunol. 2006, 177, 7497–7504.
- Lang, R.; Raffi, F.A.M. Dual-Specificity Phosphatases in Immunity and Infection: An Update. Int. J. Mol. Sci. 2019, 20, 2710.
- Theodosiou, A.; Ashworth, A. MAP Kinase Phosphatases. Genome Biol. 2002, 3, REVIEWS3009.
- Jeong, D.G.; Yoon, T.S.; Jung, S.K.; Park, B.C.; Park, H.; Ryu, S.E.; Kim, S.J. Exploring Binding Sites Other than the Catalytic Core in the Crystal Structure of the Catalytic Domain of MKP-4. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 25–31.
- Hong, S.B.; Lubben, T.H.; Dolliver, C.M.; Petrolonis, A.J.; Roy, R.A.; Li, Z.; Parsons, T.F.; Li, P.; Xu, H.; Reilly, R.M.; et al. Expression, Purification, and Enzymatic Characterization of the Dual Specificity Mitogen-Activated Protein Kinase Phosphatase, MKP-4. Bioorg. Chem. 2005, 33, 34–44.
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437.
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.-K.; Amini, B.; Andersen, E.; Andersson, A.-C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A Human Protein Atlas for Normal and Cancer Tissues Based on Antibody Proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932.
- Xu, H.; Dembski, M.; Yang, Q.; Yang, D.; Moriarty, A.; Tayber, O.; Chen, H.; Kapeller, R.; Tartaglia, L.A. Dual Specificity Mitogen-Activated Protein (MAP) Kinase Phosphatase-4 Plays a Potential Role in Insulin Resistance. J. Biol. Chem. 2003, 278, 30187–30192.
- Jiapaer, Z.; Li, G.; Ye, D.; Bai, M.; Li, J.; Guo, X.; Du, Y.; Su, D.; Jia, W.; Chen, W.; et al. LincU Preserves Naive Pluripotency by Restricting ERK Activity in Embryonic Stem Cells. Stem Cell Rep. 2018, 11, 395–409.
- Imajo, M.; Kondoh, K.; Yamamoto, T.; Nakayama, K.; Nakajima-Koyama, M.; Nishida, E. Antagonistic Interactions between Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase and Retinoic Acid Receptor Signaling in Colorectal Cancer Cells. Mol. Cell. Biol. 2017, 37.
- Chakravarthi, V.P.; Ratri, A.; Masumi, S.; Borosha, S.; Ghosh, S.; Christenson, L.K.; Roby, K.F.; Wolfe, M.W.; Rumi, M.A.K. Granulosa Cell Genes That Regulate Ovarian Follicle Development beyond the Antral Stage: The Role of Estrogen Receptor β. Mol. Cell. Endocrinol. 2021, 528, 111212.