Polyploidy, also known as whole-genome amplification, is a condition in which the organism has more than two basic sets of chromosomes. Polyploidy frequently arises during tissue development and repair, and in age-associated diseases, such as cancer. Its consequences are diverse and clearly different between systems. The liver is a particularly fascinating organ in that it can adapt its ploidy to the physiological and pathological context.
- liver
- hepatocytes
- polyploidy
- cell cycle
- centrosome
- DNA damage response
- hepatocellular carcinoma
1. Introduction
Eukaryotic organisms usually contain two complete haploid sets of homologous chromosomes (diploidy, 2 n). Polyploidy, or whole-genome duplication, is the condition in which an organism or cell contains additional sets of chromosomes (e.g., 4 n, 8 n). These additional sets of chromosomes may originate from a single species (autopolyploidy) or from different, generally closely related, species (allopolyploidy). In polyploid cells, the number of chromosome may be amplified within a single nucleus (mononucleate polyploid cells), defining the nuclear ploidy, or the genetic material may be distributed between two or more nuclei (e.g., binucleate polyploid cells), defining cellular ploidy. It is important to distinguish between polyploidy and aneuploidy, which is a deviation from a multiple of the chromosome number in which one or more of the chromosomes are missing or present in excess.
Polyploidy was first identified more than a century ago and is considered to be an evolutionary adaptation to environmental changes [1,2][1][2]. Polyploidy is confined to certain taxa and is very common in plants and in certain groups of fish and amphibians [3,4][3][4]. However, many species that are currently diploid, including humans, are derived from polyploid ancestors [5,6][5][6]. These species, who experienced ancient genome duplications followed by genome reduction, are known as paleopolyploids. The polyploidization of an entire organism is rare in mammals, in which it generally results in lethality, spontaneous abortions or embryonic resorptions [7,8][7][8]. However, the theory that polyploidy was impossible in mammals was overturned by the discovery of tetraploidy in the red vizcacha rat and its cousin, the golden vizcacha rat [9]. Whole-organism polyploidy may be rare, but polyploid cells are present at relatively high frequency in many mammalian tissues [10,11,12,13][10][11][12][13]. In physiological conditions, the transition of cells from diploidy to polyploidy is part of the developmental program in some tissues [10,14][10][14]. Polyploid cells are found in the heart (cardiomyocytes, up to 4 n), bone marrow (megakaryocytes, up to 128 n), placenta (trophoblast giant cells, up to 64 n) and liver (hepatocytes, up to 16 n). In adult tissues, injury and cellular stress can induce cell losses and tissue homeostasis is dependent on the compensation of this cell death. Polyploidization constitutes an alternative cell loss compensation strategy in postmitotic tissues lacking stem cells [12,13,15][12][13][15]. Compensatory proliferation mechanisms have been studied in detail in Drosophila. Losick and coworkers found that the adult abdominal epidermis responds to wounding by inducing the formation of large cells, facilitating the rapid re-establishment of an epithelial barrier [16]. Polyploidization also occurs in a sizeable proportion of human tumors [10,17,18][10][17][18]. Most tumors have polyploid or near-polyploid karyotypes [19,20][19][20]. Polyploidy has been shown to be associated with tumor progression, in particular due to its association with the development of chromosome instability (CIN) [21,22,23][21][22][23].
2. Liver Polyploidy and Chronic Liver Disease
The most common causes of chronic liver disease (CLD) are chronic hepatitis B or C virus (HBV/HCV) infections, alcohol abuse and non-alcoholic fatty liver disease (NAFLD) [80][24]. Compensatory proliferation during CLD is now known to be a major cellular process leading to polyploid adaptations and transformation events. Hepatocytes retain a unique ability to proliferate rapidly in response to aggression [81][25]. A continuous cycle of hepatocyte death and compensatory hepatocyte proliferation takes place during CLD. Compensatory proliferation in CLD has also been linked to polyploid adaptations because of cell-cycle defects leading to mitotic escape. For example, human liver parenchyma infected with HBV or HCV presents an increase in the polyploid hepatocyte fraction (mononucleate and binucleate) and a parallel decrease in the diploid fraction, correlated with disease severity [52,82][26][27]. During HBV infection, the HBx protein seems to play an essential role in destabilizing the hepatocyte polyploid state. In a transgenic mouse model of HBV, HBx protein production in hepatocytes has been shown to modify the timing of the cell cycle, accelerating entry into S-phase and delaying the G2-M transition [83,84][28][29]. This phenomenon triggers a transient disruption of hepatocyte polyploidization, leading to an increase in the proportion of mononucleate hepatocytes (≥4 n) at the expense of binucleate hepatocytes [84,85][29][30]. Alteration of ploidy content in HBV infected hepatocytes has been correlated with high level of DNA damage and the activation of the mitotic kinase PLK1 (Polo Like Kinase 1) [84,85][29][30]. An increase in the proportion of polyploid cells after HCV infection has been observed in human primary hepatocytes [86][31]. It has been shown, in hepatic cells (primary hepatocytes and HepG2 and Huh7 cells), that the core proteins of HCV can induce polyploidization by impairing the spindle assembly checkpoint function, thereby promoting mitotic segregation defects. This endomitosis process has been associated to a reduced Rb transcription and enhanced E2F-1 and Mad2 expression which results in uncoupling of mitotic checkpoint [86][31]. Finally, the best characterized mechanism linking compensatory proliferation and polyploidization has been described in NAFLD. Of note, NAFLD is the most common cause of chronic liver disease worldwide it has been predicted that it will become the most common underlying etiological risk factor for HCC and liver transplantation in the future [87,88,89][32][33][34]. We investigated the ploidy profiles during the setting of this disease [90,91][35][36]. We have observed an enrichment in tetraploid and highly polyploid mononucleate hepatocytes (≥8 n) in the fatty liver parenchyma of various NAFLD mouse models (high-fat diet, ob/ob, methionine-choline-deficient diet and PTEN KO ) and in NASH patients. Using primary hepatocyte cultures from mouse models, we have shown that steatotic hepatocytes progress through the G1 phase, entering S phase, but that their progression through the S and G2 phases occurs later than in control hepatocytes. This delay was associated with activation of the “G2–M checkpoint” (the DNA damage response or DDR) controlled by the ATR–p53–p21 pathway, which precludes activation of the mitotic kinase CDK1–cyclin B. This mechanism leads to an endoreplication cycle and the abortion of mitosis. This work identified oxidative stress as a key driver of pathological polyploidization. NAFLD mouse models treated with an antioxidant present lower level of oxidative stress and pathological polyploidization. This work strongly suggested that the DNA damage induced by oxidative stress is the main driving factor underlying polyploidization. Interestingly, oxidative stress-induced DNA damage has been clearly identified as a prevalent signal for the induction of polyploidization in cardiomyocytes and giant granuloma macrophages [92,93][37][38]. Further studies are required to improve our understanding of the function of the hepatocyte polyploidy induced by the DDR in CLD, and to determine whether the polyploid contingent can subvert the DDR.
3. Polyploidy and Centrosome Amplification
The centrosome is the major microtubule-organizing center involved in polarity, migration and cell division [94][39]. In diploid cells, the centrosomes are a pair of centrioles embedded in a complex proteinaceous structure, the pericentriolar material (PCM). Centrosomes duplicate only once during S phase, to ensure that the cell carries two centrosomes at the onset of mitosis, enabling it to form a bipolar spindle, thereby guaranteeing balanced chromosomal segregation [95][40]. Polyploid cells contain extra centrosomes. More than a century ago, Boveri suggested that increases in the number of centrosomes caused cancer. It has now been clearly demonstrated that extra centrosomes cause chromosome instability, aneuploidy and trigger spontaneous tumorigenesis in multiple tissues [96,97,98][41][42][43]. Polyploid cells with extra centrosomes can undergo mitoses. These centrosomes may cluster together, acting as two single units mimicking a bipolar spindle, or may act as single entities, generating multipolar spindles [21,99][21][44]. Chromosome segregation errors are common during multipolar cell division and lead to random gains and losses of whole chromosome (aneuploidy). In the liver parenchyma, proliferating polyploid hepatocytes can form multipolar spindles; cell division then leads to the generation of progenies of lower ploidy, sometimes associated with aneuploidy, in a phenomenon known as “ploidy reduction” [50,100,101][45][46][47]. Ploidy reduction co-exists with polyploidization and re-polyploidization in injured livers [50,72][45][48]. The degree of aneuploidy in the healthy liver is still a matter of discussion [50,78,102][45][49][50]. However, it has been clearly demonstrated that ploidy reduction acts as an adaptation mechanism, enabling the liver to cope with chronic injury [72,78,103][48][49][51].
In different tissues, polyploidy is frequently associated with CIN, aneuploidy, cell transformation and tumor formation, and a number of important studies have tried to identify mechanism limiting the proliferation of polyploid cells. Pioneering studies demonstrated that tetraploidization leads to the stabilization of p53, resulting in cell cycle arrest [104,105,106][52][53][54]. Aneuploidization following tetraploidization is mostly observed in cells in which p53 is inactivated [76,107][55][56]. David Pellman’s group has shown that tetraploid, but not diploid, mammary epithelial cells in which p53 is inactivated give rise to malignant tumors in nude mice [107][56]. It is becoming clear that an aberrant number of centrosomes can trigger an antiproliferative signal in polyploid cells. Two mechanisms related to centrosome amplification and p53 activation have been reported to restrain the proliferation of the polyploid contingent. The first mechanism was deduced from the observation that large tumor suppressor kinase 2 (LATS2), the core kinase of the Hippo pathway, can be translocated from the centrosome to the nucleus, where it stabilizes p53 by inhibiting MDM2 [108][57]. David Pellman’s group demonstrated that cytokinesis failure, generating tetraploid hepatocytes, is a physiological activator of LATS2. LATS2 activates the p53 pathway in tetraploid hepatocytes, but it also inactivates YAP/TAZ-dependent transcription, thereby limiting proliferation [76][55]. Another mechanism for p53 activation in response to polyploidization was recently described. This mechanism involves a multi-protein complex, the PIDDosome. This complex was identified in 2004 as a protein complex involved in the activation of caspase-2 (CASP2) in response to genotoxic stress [109][58]. Since, it has been demonstrated that the PIDDosome, consisting of p53-induced death domain protein 1 (PIDD1), the adaptor protein RAIDD, and CASP2, acts as a molecular sensor of extra centrosomes, inducing p53-mediated cell cycle arrest if supernumerary centrosomes are detected [110][59]. PIDD1 localizes to the mother centriole. The accumulation of additional centrosomes induces assembly of the PIDDosome, through the recruitment of RAIDD and CASP2 to PIDD1. CASP2 is then activated and cleaves the E3 ubiquitin ligase MDM2, leading to p53 stabilization and p21-induced cell-cycle arrest [110][59]. This mechanism has been demonstrated in both cancer and non-transformed cells in which cytokinesis has failed [110][59]. More recently, Saldky showed, in a very elegant study, that the PIDDosome is involved in the regulation of p53 activation to limit hepatocyte polyploidy [111][60]. They showed that the PIDDosome-p53-p21 axis is activated in hepatocytes, in which it controls the extent of polyploidization during postnatal liver development. The PIDDosome is activated after the first round of cytokinesis failure in hepatocytes. E2F-family members regulate the production of CASP2 and PIDD1 for liver ploidy control. Interestingly, polyploid hepatocytes also engage the PIDDosome during the regeneration process, to prevent excessive polyploidization [111][60].
4. The Fate of Polyploid Hepatocytes during Liver Tumorigenesis
The occurrence of polyploid cells is a frequent signature of cancers [112,113][61][62] and whole-genome duplication in tumor tissues is strongly associated with copy number aberrations and a poor prognosis [20,114][20][63]. As previously described, several studies have shown that polyploid cancer cells emerge before the onset of aneuploidy and during the transition from premalignant to malignant lesions [26,115][64][65]. These findings strongly suggest that polyploid cells are genetically unstable and favor tumorigenesis [10,17][10][17]. The liver is physiologically polyploid, has to cope with a certain degree of aneuploidy and modifies it ploidy content following tissue injury and stress [24,101][66][47]. Deciphering the role of polyploidization in liver tumorigenesis is a major issue. Several studies have shown that human hepatocarcinomas (HCCs) are highly enriched in diploid hepatocytes, suggesting that polyploid status may protect against HCC and that diploid hepatocytes are the dominant drivers of tumorigenesis [116,117,118][67][68][69]. Until recently, the lack of an experimental model for manipulating ploidy levels in vivo without adverse effects for liver homeostasis hindered efforts to dissociate the tumorigenic potentials of diploid and polyploid hepatocytes. However, in recent years, great efforts have been made to develop genetic models with either low or high levels of ploidy, for careful dissection of the role of polyploidy in hepatocarcinogenesis. The groups of Leone [119][70] and Duncan [78][49] have studied the tumorigenesis process in DEN-treated E2f7/E2f8 knockout mice. They demonstrated that these mice, in which the liver is mostly diploid, are much more susceptible to transformation than control mice, the livers of which contain a mixture of diploid and polyploid hepatocytes [78,119][49][70]. Consistent with these findings, Sladky et al., observed that PIDDosome-knockout mice with a high degree of polyploidy are protected against DEN-induced HCC, and that tumor progression in these mice seems to be driven by lower-ploidy hepatocytes [111,120][60][71]. These data provide solid evidence that the polyploid state protects the liver against tumor formation. Consistent with this conclusion, E2f7/E2f8 transcripts [121][72] and some components of the PIDDosome pathway [120][71] have been reported to be strongly expressed in human HCC and positively correlated with a poor prognosis for patients.
What, then, are the intrinsic properties of polyploid hepatocytes responsible for this protection against liver tumorigenesis? Polyploid hepatocytes proliferate less efficiently than diploid hepatocytes in response to proliferative stimuli, and this may enable polyploid cells to suppress tumor development [122][73]. Zhu and coworkers proposed a number of other possibilities, in their work on mouse models harboring a hyperploid liver, in which they manipulated the weaning period [123][74] or transiently knocked down levels of the actin-binding protein anillin [123[74][75][76],124,125], a key factor required for cytokinesis [126][77]. They then explored the outcome of these hyperploid livers compared to diploid ones (loss of E2F7/8 ) in diverse cancer models. Consistent with the findings of other studies, they confirmed that mostly diploid livers were highly susceptible to HCC formation and formed abundant tumors, whereas highly polyploid livers were much less susceptible and developed few tumors [123,125][74][76]. Diploid and polyploid livers were equally susceptible to oncogene-induced carcinogenesis, but polyploid livers were more resistant to loss of heterozygosity (LOH) mechanisms mediating transformation [123][74]. In this context, we can speculate that the loss of one or more tumor suppressor genes by LOH in a diploid hepatocyte may favor tumorigenesis, whereas the gene redundancy inherent to polyploid genomes may prevent the initiation of HCC by providing a larger reservoir of tumor suppressor genes ( Figure 31 ). Together, these studies support the notion that higher levels of polyploidy suppress liver tumors by limiting both loss of heterozygosity and proliferation, two of the potential drivers of transformation.
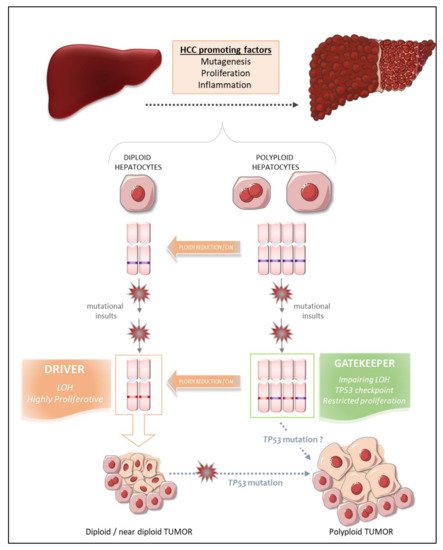
Matsumoto et al. [72][48] and Lin et al. [125][76] recently reshuffled the deck and added a level of complexity to the link between polyploidy and tumorigenesis, by revealing that polyploid hepatocytes can promote tumor initiation through ploidy reduction ( Figure 31 ) [127][78]. Contrary to the studies described above, the work of these two groups directly addressed the issue of the oncogenic potential of WT diploid and polyploid hepatocytes without the genetic manipulation of ploidy status in hepatocytes. Grompe’s group developed a new multicolor reporter allele system for genetically labeling cells, to facilitate the tracing of polyploid hepatocyte fate in vivo: heterozygous Rosa-Confetti multicolor reporter mice [72,128][48][79]. In this model, diploid cells can express only one reporter, whereas polyploid cells can be labeled by the co-expression of multiple reporters of different colors, making it easy to distinguish and follow the behavior of each cell population and its progenies. They demonstrated that polyploid hepatocytes undergo a strong decrease in ploidy in response to proliferative stimuli and that polyploid hepatocytes and their ploidy-reduced daughters play a major role in the regeneration process in chronically injured livers. The regenerative nodules emerging from proliferating polyploid hepatocytes harbored chromosomal aberrations, suggesting that ploidy reduction promoted chromosome mis-segregation and chromosomal instability (CIN). Contrary to previous reports, the authors showed, in various models of hepatocarcinogenesis (oncogenic insult or tumor-prone chronic injuries), that polyploid hepatocytes per se and their ploidy-reduced daughter cells can give rise to tumors, although the frequency was very slightly lower than that of their diploid counterparts [128][79]. Polyploid hepatocytes undergoing continuous proliferation during serial transplantation experiments were less responsive to multipolar mitosis and, consequently, to ploidy reduction through the disappearance of their supernumerary centrosomes, than their naïve untransplanted counterparts. Serially transplanted polyploid hepatocytes had a significantly lower tumorigenic potential than these “naïve” cells. Differences in the intrinsic oncogenic properties of “naïve” and transplanted polyploid hepatocytes cannot be rule out, but these data nevertheless support the emerging hypothesis that ploidy reduction may be a step-driver in tumorigenesis [128][79]. Furthermore, competitive tumor formation assays have revealed that ploidy reduction during the early steps of carcinogenesis plays a greater role in models of tumorigenesis induced by tumor suppressor loss than in oncogene-induced cancer models [128][79]. Overall, these data highlight the possibility that proliferating polyploid hepatocytes engaged in a ploidy reduction mechanism may provide a reservoir of low-ploidy cells susceptible to both LOH and CIN, two processes widely implicated in malignant transformation [129,130][80][81]. Similarly, Lin and coworkers highlighted the potent role of ploidy reduction in liver tumorigenesis [131][82]. They revealed that the hyperpolyploidization of centrilobular (CL) hepatocytes after DEN exposure was associated with the emergence of premalignant lesions in the centrilobular region. Pathological polyploidization was induced by a mechanism of abscission failure dependent on upregulation of the Aurora B kinase, a master regulator of the final step of cytokinesis [132][83]. The prevention of DEN-induced hyperpolyploidization by the inhibition of Aurora B kinase reduced the number of preneoplastic tumor foci. Finally, as these premalignant lesions were composed of significantly smaller hepatocytes, and multipolar mitoses, which can mediate genome reduction, were observed in CL hepatocyte-derived tumor cells, the authors speculated that the phenomenon of ploidy reduction sustained the transition from hyperpolyploid hepatocytes to tumor-forming cells.
Experimental studies in mouse models can provide considerable insight into the processes linking polyploidy to tumorigenesis, as data can be obtained at both the precancerous and cancerous stages. By contrast, the opportunities for dynamic investigations of the role of ploidy in human liver HCC are quite limited. Nevertheless, some groups have carefully analyzed ploidy status in tumor tissues and the surrounding non-tumoral tissue in cohorts of patients with HCC of various etiologies, to determine whether ploidy status is a potent prognostic marker of human HCC [53,120][84][71]. We have used an in-situ imaging approach to evaluate ploidy status directly in human liver parenchyma [53][84]. We first showed that, in normal liver, hepatocyte polyploidy is mostly cellular (binucleate cells). No specific zonation of the polyploid contingent was observed across the lobules. We found that the binucleate polyploid fraction decreased considerably during human liver tumorigenesis, whereas the mononucleate polyploid fraction greatly increased. These results provided the first evidence to suggest that hepatocarcinogenesis can drive a conversion from physiological cellular polyploidy to pathological nuclear polyploidy. A decrease in the proportion of binucleate hepatocytes was observed in both the tumoral and surrounding non-tumoral tissues, suggesting that decreases in cellular ploidy may be a good marker of human premalignant liver parenchyma. We then carefully analyzed the nuclear ploidy profile, considering specific histological and molecular features of HCC tumors. We observed a huge increase in the percentage of 4 n and ≥8 n mononucleate hepatocytes in poorly differentiated HCC. Furthermore, taking into account the most frequent mutations in HCC, we showed that HCCs with TP53 mutations were enriched in highly polyploid hepatocytes relative to HCCs with mutations of the TERT promoter or CTNNB1 HCC. Finally, we found that the most aggressive HCCs, particularly those with TP53 mutations, were enriched in highly polyploid hepatocytes. Together, these results support the hypothesis that polyploidization drives HCC development, at least in the context of TP53 mutation, and that the nuclear ploidy spectrum can be used as a marker of HCC aggressiveness. By contrast, Sladky et al. published results suggesting that polyploidy attenuates hepatocarcinogenesis in humans. They investigated ploidy status in HCC patients by assuming that cell density (i.e., the number of cells per field) would be a good indicator of ploidy: lower-ploidy hepatocytes are small and their density is high in the liver, whereas polyploid hepatocytes are large and found at lower density [120,133][71][85]. They found that cell density in tumoral tissue tended to be lower than that in the surrounding liver and that low ploidy levels in HCC tumors (high cell density) were associated with better recurrence-free survival [120][71]. The conclusions of these two studies diverge, but the discrepancies may be explained by the functional status of TP53. Indeed, in a context of TP53 mutation, polyploid hepatocytes may be able to undergo unrestricted proliferation and multipolar mitoses due to the presence of supernumerary centrosomes, facilitating the emergence of genomic aberrations/CIN, rendering the tumor more aggressive ( Figure 31 ) [53,128][84][79]. Conversely, the presence of a functional TP53 would tend to attenuate this effect [120][71]. Hepatocarcinogenesis is a multistep process dependent on interplay between various factors, including risk factors, genetic factors and microenvironment factors, driving the formation of HCCs with considerable heterogeneity [134][86]. The assignment of a reliable and unambiguous prognostic value to ploidy status in human HCC will therefore require additional studies carefully integrating the genetic, molecular, etiological and histological landscapes of each individual tumor, and the heterogeneity within and between tumors.
References
- Comai, L. The Advantages and Disadvantages of Being Polyploid. Nat. Rev. Genet. 2005, 6, 836–846.
- Otto, S.P.; Whitton, J. Polyploid Incidence and Evolution. Annu. Rev. Genet. 2000, 34, 401–437.
- Soltis, D.E.; Bell, C.D.; Kim, S.; Soltis, P.S. Origin and Early Evolution of Angiosperms. Ann. N. Y. Acad. Sci. 2008, 1133, 3–25.
- Mable, B.; Alexandrou, M.A.; Taylor, M.I. Genome Duplication in Amphibians and Fish: An Extended Synthesis. J. Zool. 2011, 284, 151–182.
- Van de Peer, Y.; Maere, S.; Meyer, A. The Evolutionary Significance of Ancient Genome Duplications. Nat. Rev. Genet. 2009, 10, 725–732.
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 2017, 18, 411–424.
- Creasy, M.R.; Alberman, E.D. Congenital Malformations of the Central Nervous System in Spontaneous Abortions. J. Med. Genet. 1976, 13, 9–16.
- Kaufman, M.H. Histochemical Identification of Primordial Germ Cells and Differentiation of the Gonads in Homozygous Tetraploid Mouse Embryos. J. Anat. 1991, 179, 169–181.
- Gallardo, M.H.; Bickham, J.W.; Honeycutt, R.L.; Ojeda, R.A.; Köhler, N. Discovery of Tetraploidy in a Mammal. Nature 1999, 401, 341.
- Davoli, T.; de Lange, T. The Causes and Consequences of Polyploidy in Normal Development and Cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610.
- Fox, D.T.; Duronio, R.J. Endoreplication and Polyploidy: Insights into Development and Disease. Development 2013, 140, 3–12.
- Ovrebo, J.I.; Edgar, B.A. Polyploidy in Tissue Homeostasis and Regeneration. Development 2018, 145, dev156034.
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14.
- Pandit, S.K.; Westendorp, B.; de Bruin, A. Physiological Significance of Polyploidization in Mammalian Cells. Trends Cell Biol. 2013, 23, 556–566.
- Losick, V.P. Wound-Induced Polyploidy Is Required for Tissue Repair. Adv. Wound Care 2016, 5, 271–278.
- Losick, V.P.; Fox, D.T.; Spradling, A.C. Polyploidization and Cell Fusion Contribute to Wound Healing in the Adult Drosophila Epithelium. Curr. Biol. 2013, 23, 2224–2232.
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, Aneuploidy and Cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162.
- Lens, S.M.A.; Medema, R.H. Cytokinesis Defects and Cancer. Nat. Rev. Cancer 2019, 19, 32–45.
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121.
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.-Z.; Wala, J.; Mermel, C.H.; et al. Pan-Cancer Patterns of Somatic Copy Number Alteration. Nat. Genet. 2013, 45, 1134–1140.
- Storchova, Z.; Pellman, D. From Polyploidy to Aneuploidy, Genome Instability and Cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54.
- Nguyen, H.G.; Ravid, K. Polyploidy: Mechanisms and cancer promotion in hematopoietic and other cells. In Polyploidization and Cancer; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2010; pp. 105–122. ISBN 978-1-4419-6198-3.
- Wangsa, D.; Braun, R.; Schiefer, M.; Gertz, E.M.; Bronder, D.; Quintanilla, I.; Padilla-Nash, H.M.; Torres, I.; Hunn, C.; Warner, L.; et al. The Evolution of Single Cell-Derived Colorectal Cancer Cell Lines Is Dominated by the Continued Selection of Tumor Specific Genomic Imbalances, Despite Random Chromosomal Instability. Carcinogenesis 2018, 39, 993–1005.
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the Prevalence of the Most Common Causes of Chronic Liver Diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530.
- Magami, Y.; Azuma, T.; Inokuchi, H.; Kokuno, S.; Moriyasu, F.; Kawai, K.; Hattori, T. Cell Proliferation and Renewal of Normal Hepatocytes and Bile Duct Cells in Adult Mouse Liver. Liver 2002, 22, 419–425.
- Toyoda, H.; Bregerie, O.; Vallet, A.; Nalpas, B.; Pivert, G.; Brechot, C.; Desdouets, C. Changes to Hepatocyte Ploidy and Binuclearity Profiles during Human Chronic Viral Hepatitis. Gut 2005, 54, 297–302.
- Toyoda, H.; Kumada, T.; Bregerie, O.; Brechot, C.; Desdouets, C. Conserved Balance of Hepatocyte Nuclear DNA Content in Mononuclear and Binuclear Hepatocyte Populations during the Course of Chronic Viral Hepatitis. World J. Gastroenterol. 2006, 12, 4546–4548.
- Rakotomalala, L.; Studach, L.; Wang, W.-H.; Gregori, G.; Hullinger, R.L.; Andrisani, O. Hepatitis B Virus X Protein Increases the Cdt1-to-Geminin Ratio Inducing DNA Re-Replication and Polyploidy. J. Biol. Chem. 2008, 283, 28729–28740.
- Studach, L.; Wang, W.-H.; Weber, G.; Tang, J.; Hullinger, R.L.; Malbrue, R.; Liu, X.; Andrisani, O. Polo-like Kinase 1 Activated by the Hepatitis B Virus X Protein Attenuates Both the DNA Damage Checkpoint and DNA Repair Resulting in Partial Polyploidy. J. Biol. Chem. 2010, 285, 30282–30293.
- Ahodantin, J.; Bou-Nader, M.; Cordier, C.; Mégret, J.; Soussan, P.; Desdouets, C.; Kremsdorf, D. Hepatitis B Virus X Protein Promotes DNA Damage Propagation through Disruption of Liver Polyploidization and Enhances Hepatocellular Carcinoma Initiation. Oncogene 2019, 38, 2645–2657.
- Machida, K.; Liu, J.-C.; McNamara, G.; Levine, A.; Duan, L.; Lai, M.M.C. Hepatitis C Virus Causes Uncoupling of Mitotic Checkpoint and Chromosomal Polyploidy through the Rb Pathway. J. Virol. 2009, 83, 12590–12600.
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922.
- Younossi, Z.M. The Epidemiology of Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 11, 92–94.
- Gentric, G.; Desdouets, C. Liver Polyploidy: Dr Jekyll or Mr Hide? Oncotarget 2015, 6, 8430–8431.
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative Stress Promotes Pathologic Polyploidization in Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2015, 125, 981–992.
- Herrtwich, L.; Nanda, I.; Evangelou, K.; Nikolova, T.; Horn, V.; Erny, D.; Stefanowski, J.; Rogell, L.; Klein, C.; Gharun, K.; et al. DNA Damage Signaling Instructs Polyploid Macrophage Fate in Granulomas. Cell 2016, 167, 1264–1280.
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 2014, 157, 565–579.
- Bettencourt-Dias, M.; Glover, D.M. Centrosome Biogenesis and Function: Centrosomics Brings New Understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463.
- Nigg, E.A. Centrosome Duplication: Of Rules and Licenses. Trends Cell Biol. 2007, 17, 215–221.
- Levine, M.S.; Holland, A.J. The Impact of Mitotic Errors on Cell Proliferation and Tumorigenesis. Genes Dev. 2018, 32, 620–638.
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A Mechanism Linking Extra Centrosomes to Chromosomal Instability. Nature 2009, 460, 278–282.
- Godinho, S.A.; Pellman, D. Causes and Consequences of Centrosome Abnormalities in Cancer. Philos. Trans. R. Soc. B. 2014, 369, 20130467.
- Duensing, A.; Duensing, S. Centrosomes, Polyploidy and Cancer. Adv. Exp. Med. Biol. 2010, 676, 93–103.
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The Ploidy Conveyor of Mature Hepatocytes as a Source of Genetic Variation. Nature 2010, 467, 707–710.
- Duncan, A.W.; Hickey, R.D.; Paulk, N.K.; Culberson, A.J.; Olson, S.B.; Finegold, M.J.; Grompe, M. Ploidy Reductions in Murine Fusion-Derived Hepatocytes. PLoS Genet. 2009, 5, e1000385.
- Duncan, A.W. Aneuploidy, Polyploidy and Ploidy Reversal in the Liver. Semin. Cell Dev. Biol. 2013, 24, 347–356.
- Matsumoto, T.; Wakefield, L.; Tarlow, B.D.; Grompe, M. In Vivo Lineage Tracing of Polyploid Hepatocytes Reveals Extensive Proliferation during Liver Regeneration. Cell Stem Cell 2020, 26, 34–47.
- Wilkinson, P.D.; Alencastro, F.; Delgado, E.R.; Leek, M.P.; Weirich, M.P.; Otero, P.A.; Roy, N.; Brown, W.K.; Oertel, M.; Duncan, A.W. Polyploid Hepatocytes Facilitate Adaptation and Regeneration to Chronic Liver Injury. Am. J. Pathol. 2019, 189, 1241–1255.
- Knouse, K.A.; Lopez, K.E.; Bachofner, M.; Amon, A. Chromosome Segregation Fidelity in Epithelia Requires Tissue Architecture. Cell 2018, 175, 200–211.
- Duncan, A.W.; Hanlon Newell, A.E.; Smith, L.; Wilson, E.M.; Olson, S.B.; Thayer, M.J.; Strom, S.C.; Grompe, M. Frequent Aneuploidy among Normal Human Hepatocytes. Gastroenterology 2012, 142, 25–28.
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid State Induces P53-Dependent Arrest of Nontransformed Mammalian Cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328.
- Meraldi, P.; Honda, R.; Nigg, E.A. Aurora-A Overexpression Reveals Tetraploidization as a Major Route to Centrosome Amplification in P53-/- Cells. EMBO J. 2002, 21, 483–492.
- Kuffer, C.; Kuznetsova, A.Y.; Storchová, Z. Abnormal Mitosis Triggers P53-Dependent Cell Cycle Arrest in Human Tetraploid Cells. Chromosoma 2013, 122, 305–318.
- Ganem, N.J.; Cornils, H.; Chiu, S.-Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Théry, M.; Camargo, F.D.; Pellman, D. Cytokinesis Failure Triggers Hippo Tumor Suppressor Pathway Activation. Cell 2014, 158, 833–848.
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis Failure Generating Tetraploids Promotes Tumorigenesis in P53-Null Cells. Nature 2005, 437, 1043–1047.
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A Positive Feedback Loop between the P53 and Lats2 Tumor Suppressors Prevents Tetraploidization. Genes Dev. 2006, 20, 2687–2700.
- Tinel, A.; Tschopp, J. The PIDDosome, a Protein Complex Implicated in Activation of Caspase-2 in Response to Genotoxic Stress. Science 2004, 304, 843–846.
- Fava, L.L.; Schuler, F.; Sladky, V.; Haschka, M.D.; Soratroi, C.; Eiterer, L.; Demetz, E.; Weiss, G.; Geley, S.; Nigg, E.A.; et al. The PIDDosome Activates P53 in Response to Supernumerary Centrosomes. Genes Dev. 2017, 31, 34–45.
- Sladky, V.C.; Knapp, K.; Soratroi, C.; Heppke, J.; Eichin, F.; Rocamora-Reverte, L.; Szabo, T.G.; Bongiovanni, L.; Westendorp, B.; Moreno, E.; et al. E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Dev. Cell 2020, 52, 335–349.
- Zasadil, L.M.; Britigan, E.M.C.; Weaver, B.A. 2n or Not 2n: Aneuploidy, Polyploidy and Chromosomal Instability in Primary and Tumor Cells. Semin. Cell Dev. Biol. 2013, 24, 370–379.
- Tanaka, K.; Goto, H.; Nishimura, Y.; Kasahara, K.; Mizoguchi, A.; Inagaki, M. Tetraploidy in Cancer and Its Possible Link to Aging. Cancer Sci. 2018, 109, 2632–2640.
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195.
- Olaharski, A.J.; Sotelo, R.; Solorza-Luna, G.; Gonsebatt, M.E.; Guzman, P.; Mohar, A.; Eastmond, D.A. Tetraploidy and Chromosomal Instability Are Early Events during Cervical Carcinogenesis. Carcinogenesis 2006, 27, 337–343.
- Maley, C.C.; Galipeau, P.C.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Reid, B.J. The Combination of Genetic Instability and Clonal Expansion Predicts Progression to Esophageal Adenocarcinoma. Cancer Res. 2004, 64, 7629–7633.
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in Liver Development, Homeostasis and Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 1–15.
- Anti, M.; Marra, G.; Rapaccini, G.L.; Rumi, C.; Bussa, S.; Fadda, G.; Vecchio, F.M.; Valenti, A.; Percesepe, A.; Pompili, M.; et al. DNA Ploidy Pattern in Human Chronic Liver Diseases and Hepatic Nodular Lesions. Flow Cytometric Analysis on Echo-Guided Needle Liver Biopsy. Cancer 1994, 73, 281–288.
- Fujimoto, J.; Okamoto, E.; Yamanaka, N.; Toyosaka, A.; Mitsunobu, M. Flow Cytometric DNA Analysis of Hepatocellular Carcinoma. Cancer 1991, 67, 939–944.
- Nagasue, N.; Kohno, H.; Hayashi, T.; Yamanoi, A.; Uchida, M.; Takemoto, Y.; Makino, Y.; Ono, T.; Hayashi, J.; Nakamura, T. Lack of Intratumoral Heterogeneity in DNA Ploidy Pattern of Hepatocellular Carcinoma. Gastroenterology 1993, 105, 1449–1454.
- Kent, L.N.; Rakijas, J.B.; Pandit, S.K.; Westendorp, B.; Chen, H.-Z.; Huntington, J.T.; Tang, X.; Bae, S.; Srivastava, A.; Senapati, S.; et al. E2f8 Mediates Tumor Suppression in Postnatal Liver Development. J. Clin. Investig. 2016, 126, 2955–2969.
- Sladky, V.C.; Knapp, K.; Szabo, T.G.; Braun, V.Z.; Bongiovanni, L.; van den Bos, H.; Spierings, D.C.; Westendorp, B.; Curinha, A.; Stojakovic, T.; et al. PIDDosome-Induced P53-Dependent Ploidy Restriction Facilitates Hepatocarcinogenesis. EMBO Rep. 2020, 21, e50893.
- Moreno, E.; Toussaint, M.J.M.; van Essen, S.C.; Bongiovanni, L.; van Liere, E.A.; Koster, M.H.; Yuan, R.; van Deursen, J.M.; Westendorp, B.; de Bruin, A. E2F7 Is a Potent Inhibitor of Liver Tumor Growth in Adult Mice. Hepatology 2021, 73, 303–317.
- Wilkinson, P.D.; Delgado, E.R.; Alencastro, F.; Leek, M.P.; Roy, N.; Weirich, M.P.; Stahl, E.C.; Otero, P.A.; Chen, M.I.; Brown, W.K.; et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatolology 2019, 69, 1242–1258.
- Zhang, S.; Zhou, K.; Luo, X.; Li, L.; Tu, H.-C.; Sehgal, A.; Nguyen, L.H.; Zhang, Y.; Gopal, P.; Tarlow, B.D.; et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev. Cell 2018, 44, 447–459.
- Zhang, S.; Nguyen, L.H.; Zhou, K.; Tiu, H.C.; Sehgal, A.; Nassour, I.; Li, L.; Gopal, P.; Goodman, J.; Singal, A.G.; et al. Knockdown of Anillin Actin Binding Protein Blocks Cytokinesis in Hepatocytes and Reduces Liver Tumor Development in Mice without Affecting Regeneration. Gastroenterology 2018, 154, 1421–1434.
- Lin, Y.-H.; Zhang, S.; Zhu, M.; Lu, T.; Chen, K.; Wen, Z.; Wang, S.; Xiao, G.; Luo, D.; Jia, Y.; et al. Mice With Increased Numbers of Polyploid Hepatocytes Maintain Regenerative Capacity but Develop Fewer Tumors Following Chronic Liver Injury. Gastroenterology 2020, 158, 1698–1712.
- Piekny, A.J.; Maddox, A.S. The Myriad Roles of Anillin during Cytokinesis. Semin. Cell Dev. Biol. 2010, 21, 881–891.
- Müller, M.; May, S.; Bird, T.G. Ploidy Dynamics Increase the Risk of Liver Cancer Initiation. Nat. Commun. 2021, 12, 1896.
- Matsumoto, T.; Wakefield, L.; Peters, A.; Peto, M.; Spellman, P.; Grompe, M. Proliferative Polyploid Cells Give Rise to Tumors via Ploidy Reduction. Nat. Commun. 2021, 12, 646.
- Vargas-Rondón, N.; Villegas, V.E.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4.
- Bach, D.-H.; Zhang, W.; Sood, A.K. Chromosomal Instability in Tumor Initiation and Development. Cancer Res. 2019, 79, 3995–4002.
- Lin, H.; Huang, Y.-S.; Fustin, J.-M.; Doi, M.; Chen, H.; Lai, H.-H.; Lin, S.-H.; Lee, Y.-L.; King, P.-C.; Hou, H.-S.; et al. Hyperpolyploidization of Hepatocyte Initiates Preneoplastic Lesion Formation in the Liver. Nat. Commun. 2021, 12, 645.
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The Chromosomal Passenger Complex (CPC): From Easy Rider to the Godfather of Mitosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 789–803.
- Bou-Nader, M.; Caruso, S.; Donne, R.; Celton-Morizur, S.; Calderaro, J.; Gentric, G.; Cadoux, M.; L’Hermitte, A.; Klein, C.; Guilbert, T.; et al. Polyploidy Spectrum: A New Marker in HCC Classification. Gut 2019, 69, 355–364.
- Duncan, A.W. Hepatocyte Ploidy Modulation in Liver Cancer. EMBO Rep. 2020, 21, e51922.
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primer 2021, 7, 6.