Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Robert Ernst.
The unfolded protein response (UPR) bears an evolutionary conserved, dual sensitivity to both protein-folding imbalances in the endoplasmic reticulum (ER) lumen and aberrant compositions of the ER membrane, referred to as lipid bilayer stress (LBS). Through transcriptional and non-transcriptional mechanisms, the UPR upregulates the protein folding capacity of the ER and balances the production of proteins and lipids to maintain a functional secretory pathway.
- UPR
- IRE1
- PERK
- ATF6
- ER
- lipid bilayer stress
1. Introduction
The endoplasmic reticulum (ER) spans eukaryotic cells as a membrane-bound organelle with functionally and structurally distinct subdomains. Its membrane includes both the nuclear envelope (NE) separating the nucleoplasm from the cytoplasm, and the peripheral ER forming an elaborate network of tubules and cisternae [1]. The ER has important functions in cellular signaling, secretion, membrane protein biogenesis, and lipid metabolism. In most eukaryotic cells, the ER acts as a storage compartment for intracellular Ca2+, which can be released into the cytosol as an important second messenger in a wealth of signaling pathways [2]. A large portion of the ER surface contributes to membrane contact sites (MCSs), which provide a physical link to other organelles for exchanging ions and small molecules such as lipids [3].
The ER is a hotspot for protein folding and membrane biogenesis [4,5][4][5]. It is the entry point to the secretory pathway and contributes to the biogenesis of peroxisomes, lipid droplets, and autophagic membranes. All proteins entering the endomembrane system via the secretory pathway are synthesized by cytosolic ribosomes. In mammalian cells, the ER handles roughly 10,000 different proteins (about 30% of the proteome), thereby accumulating between 0.1 and 2.0 million client proteins every minute in some cell types [6]. Most proteins entering the secretory pathway in Saccharomyces cerevisiae (S. cerevisiae) are membrane proteins. They are co- or post-translationally inserted into the lipid bilayer and folded with the help of molecular chaperones. Probably the most prominent chaperone in the lumen of the ER is the immunoglobulin heavy-chain binding protein (BiP; GRP78; Kar2p in S. cerevisiae) [7]. In a similar way to all HSP70 chaperones, it binds and hydrolyzes ATP in its nucleotide binding domain (NBD), whilst undergoing cycles of binding and releasing client proteins at its substrate binding domain (SBD). This dynamic interaction with unfolded proteins prevents their unfavorable interactions and co-aggregation with other folding intermediates. Nucleotide exchange factors and J-domain-containing co-chaperones (ERdj1-8) modulate the activity of BiP and help recruit clients [7,8][7][8]. When a protein fails to fold in the ER, it can be subjected to the ER-associated degradation (ERAD) machinery and degraded via the ubiquitin-proteasome system [9]. Successfully folded proteins and properly glycosylated proteins, however, can exit the ER via COPII vesicles for their transport to the Golgi apparatus [10,11][10][11].
A carefully orchestrated machine for endomembrane trafficking guarantees the distribution of soluble and membrane proteins to their final, subcellular destination. Cargo sorting relies on specific cargo receptors, but also—in the case of membrane proteins—on the physicochemical properties of the transmembrane domains [10,11,12][10][11][12]. The dynamic partitioning of a transmembrane protein between an emerging transport vesicle and its donor organelle provides a means to concentrate it in one or the other compartment. The hydrophobic thickness of a transmembrane domain and its membrane environment is particularly relevant for such mismatch-based sorting mechanisms [12,13][12][13]. A gradual increase in membrane stiffness along the secretory pathway facilitates a step-by-step sorting of transmembrane proteins with increasing hydrophobic thicknesses at each station along the secretory pathway from the ER to the plasma membrane [10,12,13,14,15][10][12][13][14][15]. In this context, the ER membrane fulfils a special role, because it has to insert, fold and assemble all sorts of transmembrane proteins, irrespective of their largely distinct transmembrane domains (on average ~20.3/20.6 hydrophobic residues in a transmembrane helix for proteins of the early secretory compared with ~24.4/27 for proteins of the late secretory pathway in mammals/fungi) [13]. To provide a suitable environment for this diverse set of membrane proteins, the ER membrane must be particularly soft and deformable; to this end, sensitive surveillance systems keep the sterol concentration in the ER low (~5–10 mol%) [16,17][16][17] and the proportion of poorly packing, mono-unsaturated fatty acyl chains high (>70 mol%) [18].
The ER is also a hotspot of lipid biosynthesis and hosts a vast repertoire of lipid metabolic enzymes [19,20][19][20]. Lipogenic enzymes in the ER include fatty acid desaturases and elongases, as well as dozens of enzymes for producing glycerophospholipids, sterols, ceramides (the precursors for more complex sphingolipids), and neutral storage lipids [19,20][19][20]. In contrast to secretory and membrane proteins, which commute between organelles via vesicular carriers, lipids are distributed also by lipid transfer proteins [3]. However, despite the continuous and rapid exchange of membrane material via vesicular and non-vesicular transport mechanisms, each organelle of the endomembrane system pathway maintains its own, characteristic membrane properties as a means to establish its identity [21,22][21][22].
2. The Conventional Role of the Unfolded Protein Response (UPR) Is Proteostasis
Cells need to adapt when the folding machinery of the ER is overwhelmed and when unfolded or misfolded proteins accumulate—a situation referred to as ER stress. The UPR was originally identified as a signaling pathway that senses ER stress to upregulate ER-resident chaperones [27,28,29][23][24][25]. Soon, it became clear that the UPR regulates not only the folding machinery in the ER, but also the processes with relevance for the entire secretory pathway, including lipid metabolism, protein translocation, ER-associated protein degradation (ERAD), ER-to-Golgi transport and Golgi-to-ER retrieval, protein glycosylation in the ER and the Golgi apparatus, vacuolar targeting, distal secretion, and cell wall biogenesis [30][26]. In mammals, the UPR relies on three single-pass, transmembrane proteins in the ER; namely, the activating transcription factor 6 (present as the isoforms ATF6α/β in mammals), the inositol-requiring enzyme 1 (with two mammalian isoforms IRE1α/β), and the double-stranded RNA-activated protein kinase (PKR)—such as ER kinase (PERK) [24][27]. IRE1 constitutes the most conserved branch of the UPR and represents the only UPR transducer in S. cerevisiae (ScIRE1).
When activated, the UPR (1) lowers the global rate of protein production, (2) upregulates the rate of membrane lipid biosynthesis, (3) induces the production of ER-luminal chaperones and components of the ERAD machinery, and (4) expands the capacity of the secretory pathway [24][27]. If these adaptive responses are insufficient to restore ER homeostasis, the prolonged activity of the UPR can lead to cell death [24][27]. Given the broad transcriptional and non-transcriptional effector functions of the UPR, its crucial role in cell fate decisions between life, death, and differentiation, is unsurprising [24,31,32,33][27][28][29][30].
The upregulation of membrane lipid biosynthesis via UPR signals in S. cerevisiae causes an expansion of the ER membrane network [34,35][31][32]. Likewise, all three branches of the mammalian UPR regulate key steps of lipid metabolism and contribute to the ER membrane expansion via transcriptional and non-transcriptional mechanisms [36,37,38,39][33][34][35][36]. How the composition and properties of the ER membrane contribute to UPR activation in return has been lagging. Meanwhile, it is clear that a variety of signals originating from the ER membrane serve as potent signals for UPR activation in lipid metabolic adaptation and disease [31,40,41][28][37][38]. It is true, for example, that insulin-producing β-cells rely on UPR signals for their normal differentiation into professional, secretory cells [42][39], but it is the chronic stress caused by the excess of saturated fatty acids that kills them [43][40]. Before going into a more detailed discussion of the signals that lead to UPR activation, we will introduce the three branches of the mammalian UPR.
3. Three Musketeers—The Mammalian UPR Transducers
ATF6 is a single-pass, type II transmembrane protein. Its C-terminal, ER-luminal domain forms intermolecular disulfide bonds that stabilize homo-oligomeric, inactive assemblies [44,45][41][42]. This way, ATF6 is directly sensitive to reducing conditions that would interfere with the normal oxidative folding in the ER. Furthermore, ATF6 is associated with the molecular ER chaperone BiP [46][43]. Under conditions of ER stress, BiP is released from ATF6 and unmasks two conserved Golgi localization signals [46][43]. BiP dissociation can be induced in vitro by supplementing ATP-Mg2+ to immuno-isolated complexes, yet additional factors such as co-chaperones are proposed to control ATF6 activation in vivo by regulating the ATPase cycle of BiP [47][44]. Following BiP dissocation, ATF6 is packaged in COPII vesicles [48][45] and transported to the Golgi apparatus, where it is proteolytically processed and activated via the site-1-protease and site-2-protease [49,50][46][47]. Released from its membrane anchor, the transcriptionally active ATF6p50 fragment enters the nucleus and triggers a broad transcriptional program to reestablish the protein folding homeostasis in the ER (Figure 1, left panel) [51,52,53][48][49][50].
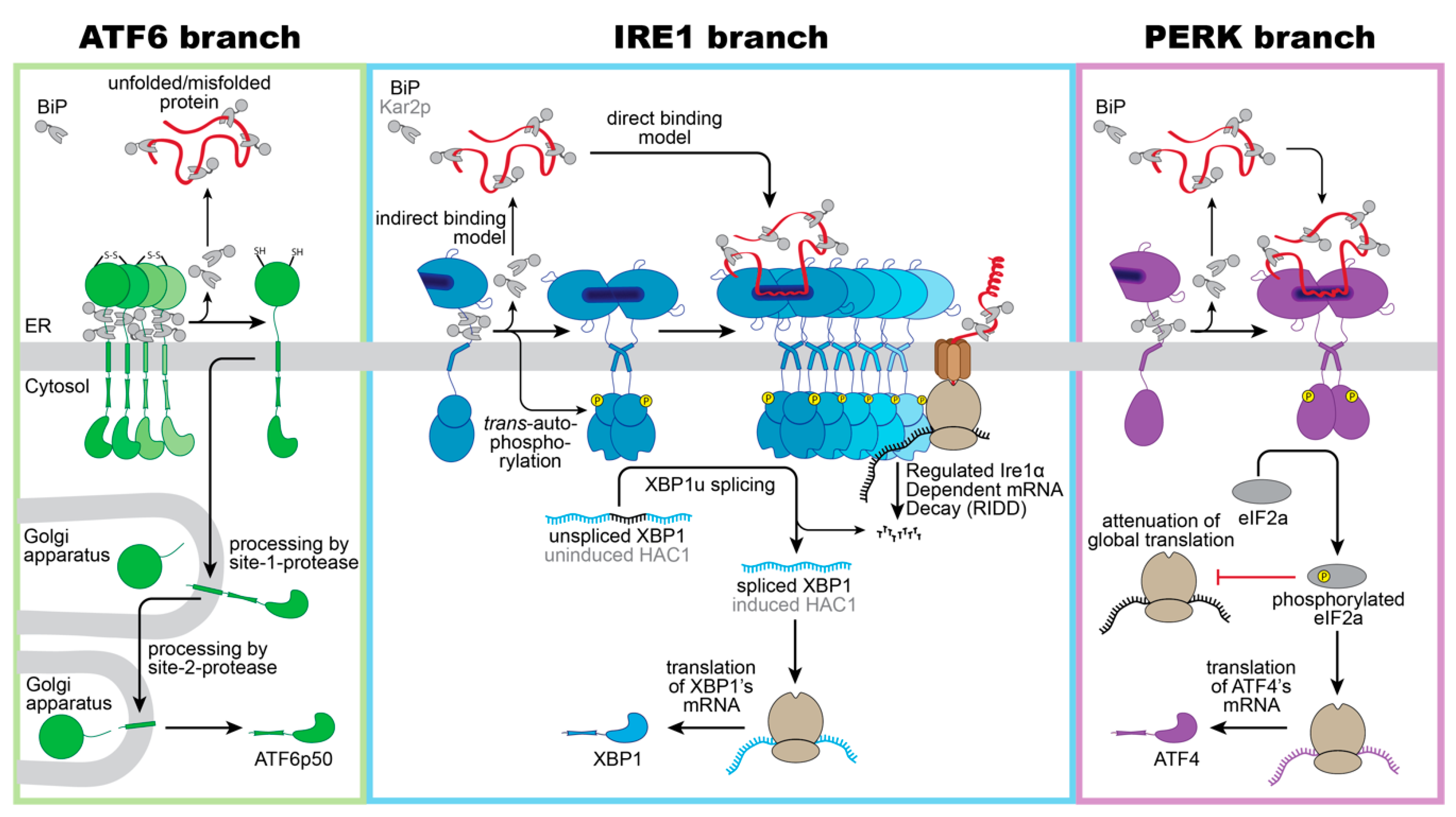
Figure 1. Schematic overview of proteotoxic ER stress signaling by the UPR. Three branches of the UPR sense proteotoxic stress in the ER to control adaptive transcriptional and non-transcriptional responses: ATF6, IRE1 (IRE1α in mammals/ ScIRE1 in S. cerevisiae) and PERK. (Left panel): Intermolecular disulfide bonds stabilize inactive homo-oligomers of ATF6 thereby limiting the pool of ATF6 that can be activated. In the absence of ER stress, ATF6 interacts with its negative regulator BiP. Notably, various regions of the ER-luminal domain of ATF6 contribute to BiP binding, but only a membrane-proximal binding region is indicated here for simplicity. Current models suggest a dissociation of BiP from ATF6 upon ER stress. The unmasking of Golgi localization signals allows for packaging of ATF6 into COPII vesicles for a transport to the Golgi apparatus. Processing by the site-1-protease and site-2-protease releases a transcriptionally active fragment (ATF6p50) for regulating UPR target genes in the nucleus. (Middle panel): Inactive monomers of IRE1α/ScIre1 associate with BiP/Kar2p via various interaction sites. Proteotoxic ER stress causes a dimerization of IRE1α/ScIre1, the release of BiP/Kar2p, and the formation of higher oligomeric assemblies of IRE1α/ScIre1. The enforced proximity of the cytosolic effector domains enables a trans-autophosphorylation of the cytosolic kinase domain and activation of the RNase domain. Oligomers of IRE1α and ScIre1 cleave the mRNA of unspliced XBP1 and uninduced HAC1, respectively, as the committed step for unconventional splicing. Translation of the spliced mRNA yields an active transcription factor XBP1/HAC1 upregulating hundreds of UPR target genes in the nucleus. Oligomers of the mammalian IRE1α oligomers can reduce the load of the ER with unfolded proteins via the regulated IRE1α-dependent mRNA Decay (RIDD). (Right panel): Proteotoxic ER stress causes a dissociation of BiP from PERK and facilitates the formation of PERK dimers and oligomers. Trans-autophosphorylation activates PERK’s cytosolic kinase domain, which then phosphorylates the eukaryotic initiation 2α (eIF2α). This causes a global attenuation of translation, but also selectively promotes the production of the transcription factor ATF4. ATF4 controls both pro-survival and pro-apoptotic signals.
A recent study suggested that the ER-resident oxidoreductase ERp18 associates with ATF6 and forms mixed disulfides specifically under conditions of ER stress to ensure optimal processing [54][51]. In fact, ERp18-depletion accelerates the rate ATF6 ER-to-Golgi trafficking, but causes aberrant processing and releases a non-productive fragment, which is not further processed by the site-2-protease [54][51].
Precisely how unfolded proteins and BiP ‘monomerize’ ATF6, despite a fully developed basic leucine zipper on the cytosolic side of the ER membrane, remains unexplored. Unlike the other two branches of the UPR, ATF6 cannot lower the flux of unfolded proteins into the ER. Instead, ATF6p50 induces an expansion of the ER membrane [37,38][34][35] and upregulates numerous genes encoding for ER chaperones, ER-luminal disulfide oxidoreductases, and ERAD components [38,51,52,53][35][48][49][50]. Notably, ATF6p50 and the transcription factor X-box-binding protein 1 (XBP1s), generated by the IRE1 branch of the UPR, act synergistically and can hetero-dimerize [51][48].
ScIRE1/IRE1α is a type I transmembrane protein conserved from yeast to humans. When unfolded proteins accumulate in the ER, ScIRE1/IRE1α oligomerizes [55][52], thereby juxtaposing the cytosolic kinase/RNase domains. This triggers the trans-autophosphorylation of the kinase domain and the activation of the RNase domain [28,56,57,58,59,60][24][53][54][55][56][57]. IRE1α excises a small intron from the XBP1 mRNA and initiates an unconventional splicing reaction, which ultimately provides a template for the transcription factor XBP1s (‘s’ stands for spliced) [59,61][56][58]. After the cleavage and ejection of the intron, the two exons of the XBP1 mRNA zipper up, form an extended stem, and become ligated by the catalytic subunit of the tRNA ligase complex RTBC [62,63][59][60]. Similarly, ScIRE1 initiates the unconventional splicing of the HAC1 mRNA for generating an active transcription factor Hac1p (Figure 1, middle panel) [57,64][54][61]. XBP1s in mammals and Hac1p in S. cerevisiae control large transcriptional programs with hundreds of target genes involved in various aspects of membrane biogenesis, protein folding, trafficking, and degradation [30,52,53,65][26][49][50][62]. IRE1α can also lower the influx of proteins by degrading mRNAs associated with the ribosome-translocon complex in a process known as IRE1-dependent mRNA decay (RIDD) [66,67][63][64]. Furthermore, the downregulation of the biogenesis of lysosome-related organelles 1 subunit 1 (Blos1) mRNA via RIDD causes an impressive clustering of lysosomes to the perinuclear region in stressed cells, which is crucial to efficiently remove protein aggregates via late endosome-mediated microautophagy [68][65].
PERK is an ER-resident, type I transmembrane kinase [69][66]. When unfolded proteins accumulate in the ER, PERK oligomerizes and its cytosolic effector domains are activated through trans-autophosphorylation [69][66]. The activated PERK phosphorylates the eukaryotic translation initiation factor 2α (eIF2α) at serine 51, thereby rapidly inhibiting the global rate of mRNA translation and lowering the flux of proteins into the ER [69,70][66][67]. The activating transcription factor 4 (ATF4) escapes this inhibition and is selectively upregulated (Figure 1, right panel) [71][68]. ATF4 upregulates genes involved in amino acid metabolism, tRNA charging, and glutathione biosynthesis [72][69], the production of the pro-apoptotic transcription factor C/EBPP homologous protein (CHOP)—also known as growth arrest—the DNA damage-inducible gene 153 (GADD153), and the growth arrest and DNA damage-inducible gene 34 (GADD34) [71,73][68][70]. CHOP/GADD153 and GADD34 orchestrate the PERK-dependent signaling output, which can be either cytoprotective or pro-apoptotic. The cytoprotective GADD34 provides a negative feedback loop that terminates the PERK-signaling downstream of eIF2α phosphorylation, by forming a complex with the protein phosphatase 1 (PP1c) [73][70]. CHOP/GADD153, on the other hand, provides pro-apoptotic signals [74][71] by inducing the expression of the death receptor 5, leading to ligand-independent signaling via Caspase 8 [75][72]. The transient activation of IRE1α during acute ER-stress, on the contrary, attenuates the death receptor 5 mRNA level via RIDD, so that two opposing UPR signals control the death receptor 5 level, and thus apoptosis [75][72].
References
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 2015, 84, 791–811.
- Carreras-Sureda, A.; Pihán, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2018, 70, 24–31.
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835.
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191.
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99.
- Hibi, T.; Dosch, H.-M. Limiting dilution analysis of the B cell compartment in human bone marrow. Eur. J. Immunol. 1986, 16, 139–145.
- Behnke, J.; Feige, M.J.; Hendershot, L.M. BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608.
- Yamamoto, Y.; Kasai, A.; Omori, H.; Takino, T.; Sugihara, M.; Umemoto, T.; Hamasaki, M.; Hatta, T.; Natsume, T.; Morimoto, R.I.; et al. ERdj8 governs the size of autophagosomes during the formation process. J. Cell Biol. 2020, 219, e201903127.
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187.
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18.
- Zanetti, G.; Pahuja, K.B.; Studer, S.; Shim, S.; Schekman, R. COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 2012, 14, 20–28.
- Herzig, Y.; Sharpe, H.J.; Elbaz, Y.; Munro, S.; Schuldiner, M. A systematic approach to pair secretory cargo receptors with their cargo suggests a mechanism for cargo selection by Erv14. PLoS Biol. 2012, 10, e1001329.
- Sharpe, H.J.; Stevens, T.J.; Munro, S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010, 142, 158–169.
- Quiroga, R.; Trenchi, A.; Montoro, A.G.; Taubas, J.V.; Maccioni, H.J.F. Short length transmembrane domains having voluminous exoplasmic halves determine retention of Type II membrane proteins in the Golgi complex. J. Cell Sci. 2013, 126, 5344–5349.
- Kaiser, H.-J.; Orlowski, A.; Rog, T.; Nyholm, T.K.M.; Chai, W.; Feizi, T.; Lingwood, D.; Vattulainen, I.; Simons, K. Lateral sorting in model membranes by cholesterol-mediated hydrophobic matching. Proc. Natl. Acad. Sci. USA 2011, 108, 16628–16633.
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008, 8, 512–521.
- Zinser, E.; Paltauf, F.; Daum, G. Sterol composition of yeast organelle membranes and subcellular distribution of enzymes involved in sterol metabolism. J. Bacteriol. 1993, 175, 2853–2858.
- Covino, R.; Ballweg, S.; Stordeur, C.; Michaelis, J.B.; Puth, K.; Wernig, F.; Bahrami, A.; Ernst, A.M.; Hummer, G.; Ernst, R. A eukaryotic sensor for membrane lipid saturation. Mol. Cell 2016, 63, 49–59.
- Ernst, R.; Ejsing, C.S.; Antonny, B. Homeoviscous adaptation and the regulation of membrane lipids. J. Mol. Biol. 2016, 428, 4776–4791.
- Holthuis, J.C.M.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57.
- Ernst, R.; Ballweg, S.; Levental, I. Cellular mechanisms of physicochemical membrane homeostasis. Curr. Opin. Cell Biol. 2018, 53, 44–51.
- Bigay, J.; Antonny, B. Curvature, lipid packing, and electrostatics of membrane organelles: Defining cellular territories in determining specificity. Dev. Cell 2012, 23, 886–895.
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.-J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464.
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206.
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756.
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258.
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086.
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338.
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181.
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794.
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423.
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536.
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41.
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636.
- Maiuolo, J.; Bulotta, S.; Verderio, C.; Benfante, R.; Borgese, N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc. Natl. Acad. Sci. USA 2011, 108, 7832–7837.
- Brewer, J.W.; Jackowski, S. UPR-Mediated membrane biogenesis in B Cells. Biochem. Res. Int. 2012, 2012, 738471.
- Le, Q.G.; Kimata, Y. Multiple ways for stress sensing and regulation of the endoplasmic reticulum-stress sensors. Cell Struct. Funct. 2021, 46, 37–49.
- Covino, R.; Hummer, G.; Ernst, R. Integrated functions of membrane property sensors and a hidden side of the unfolded protein response. Mol. Cell 2018, 71, 458–467.
- Zhang, W.; Feng, D.; Li, Y.; Iida, K.; McGrath, B.; Cavener, D.R. PERK EIF2AK3 control of pancreatic β cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006, 4, 491–497.
- El-Assaad, W.; Buteau, J.; Peyot, M.-L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic β-cell death. Endocrinology 2003, 144, 4154–4163.
- Koba, H.; Jin, S.; Imada, N.; Ishikawa, T.; Ninagawa, S.; Okada, T.; Sakuma, T.; Yamamoto, T.; Mori, K. Reinvestigation of disulfide-bonded oligomeric forms of the unfolded protein response transducer ATF6. Cell Struct. Funct. 2020, 45, 9–21.
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 1027–1043.
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 2002, 3, 99–111.
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 2005, 25, 921–932.
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780.
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799.
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364.
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376.
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.-Y.; Kaufman, R.J. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364.
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292.
- Oka, O.B.; Lith, M.; Rudolf, J.; Tungkum, W.; Pringle, M.A.; Bulleid, N.J. ER p18 regulates activation of ATF 6α during unfolded protein response. EMBO J. 2019, 38.
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118.
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039.
- Cox, J.S.; Walter, P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 1996, 87, 391–404.
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039.
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891.
- Shen, X.; Ellis, R.E.; Lee, K.; Liu, C.-Y.; Yang, K.; Solomon, A.; Yoshida, H.; Morimoto, R.; Kurnit, D.M.; Mori, K.; et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001, 107, 893–903.
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96.
- Jurkin, J.; Henkel, T.; Nielsen, A.F.; Minnich, M.; Popow, J.; Kaufmann, T.; Heindl, K.; Hoffmann, T.; Busslinger, M.; Martinez, J. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 2014, 33, 2922–2936.
- Peschek, J.; Acosta-Alvear, D.; Mendez, A.S.; Walter, P. A conformational RNA zipper promotes intron ejection during non-conventional XBP 1 mRNA splicing. EMBO Rep. 2015, 16, 1688–1698.
- Sidrauski, C.; Cox, J.S.; Walter, P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 1996, 87, 405–413.
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66.
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331.
- Acosta-Alvear, D.; Karagöz, G.E.; Fröhlich, F.; Li, H.; Walther, T.C.; Walter, P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife 2018, 7, e43036.
- Bae, D.; Moore, K.A.; Mella, J.M.; Hayashi, S.Y.; Hollien, J. Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J. Cell Biol. 2019, 218, 1118–1127.
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274.
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904.
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108.
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633.
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-Mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022.
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 2002, 7, 335–345.
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101.
More