Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Maria-del-Carmen Cardenas-Aguayo and Version 3 by Lindsay Dong.
The α-syn, encoded by the SNCA1/PARK1 gene, is a ubiquitous protein that is abundantly expressed in kidneys and blood cells, but highly enriched in the brain, particularly in the presynaptic terminals of the neocortex, hippocampus, substantia nigra (SN), thalamus, and cerebellum. Interestingly, it has been found expressed in the cytoplasm of astrocytes and oligodendrocytes in healthy individuals.
- α-synuclein
- neuroinflammation
- neurotrophic factors
1. Structure and Physiology of α-Synuclein
The α-syn, encoded by the SNCA1/PARK1 gene, is a ubiquitous protein that is abundantly expressed in kidneys and blood cells [1][37], but highly enriched in the brain, particularly in the presynaptic terminals of the neocortex, hippocampus, substantia nigra (SN), thalamus, and cerebellum. Interestingly, it has been found expressed in the cytoplasm of astrocytes and oligodendrocytes in healthy individuals [2][3][38,39]. Different scientific approaches have converged in the description of α-syn as an intrinsically disordered protein (IDP), with unusual structural properties [4][40]. Three distinct domains (Figure 1) confer dynamic structural flexibility and remarkable conformational plasticity [4][5][6][7][40,41,42,43]; (1) The N-terminal region (amino acids 1–65) confers an α-helical structure involved in lipid membranes binding and possibly promotes α-syn oligomerization; (2) The central region (amino acids 61–95) includes the NACore phosphate-binding loop, which has been implicated in the formation of amyloid fibrils and the stabilization of the pathogenic conformation of α-syn; (3) The C-terminal region (amino-acids 96–140) is associated with the major sites of metal binding and posttranslational modification, involved in modulating the protein structure, physiological functions, and toxicity.
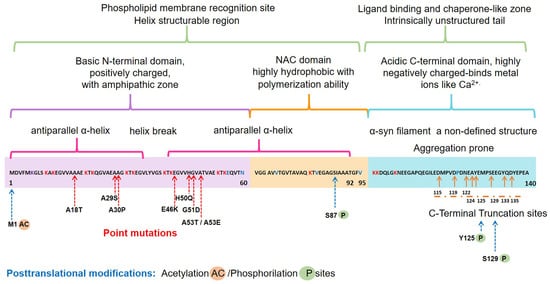
Figure 1. Schematic representation of the native α-syn monomeric structure, highlighting features linked to its biochemical function and dysfunction. Abbreviation: NAC, non-amyloid β-component.
Concerning the physiological roles of this protein, several studies have considered the subcellular localization of α-syn, which ranges from the nucleus to mitochondria and nerve terminals, to propose the following functions [8][9][10][11][12][44,45,46,47,48]: (i) neuronal health maintenance; (ii) synaptic plasticity; (iii) membrane biogenesis; iv) mitigation of oxidative stress; (v) regulation of synaptic vesicle trafficking; (vi) neurotransmitter release.
2. Pathological α-Syn Aggregates and Prion-like Properties
Several lines of evidence have proposed that native α-syn exists predominantly as an IDP monomer, which is typically found in an unfolded state and soluble in the cytosol, minimally phosphorylated in the healthy human brain. However, this dynamic protein can convert to various conformations such as helically folded tetramers resisting aggregation, pathologic oligomers, small aggregates, protofibrils, or irreversible insoluble amyloid fibrils with a stabilizing β-sheet structure [6][13][14][42,49,50]. Most α-syn forms exist in a dynamic equilibrium with each other, but perturbation of neuronal homeostasis is a starting point for pathological α-syn insolubility, self-assembly, β-sheet stacking, and misfolding. Cellular environmental cues combined with genetic factors contribute to the posttranslational modifications of the unfolded monomeric α-syn that lead to dysfunctional, neurotoxic, and pathological aggregates with a high degree of β-sheet structure [15][51].
Interestingly, all the known mutations associated with familial forms of PD are clustered within the N-terminal region, causing misfolding and/or aggregation of the mutant α-syn [6][42]. Furthermore, N-terminal acetylation could be critical for both aggregation and function [16][52]. In the C-terminal region, posttranslational modifications have been described that promote a tendency to protein aggregation. Examples are the C-terminal truncation, which results in increased filament assembly, and the phosphorylation at S129 (pS129), which regulates membrane-binding and enhances interactions with metal ions and other proteins (Figure 1). Highlighting that pS129 α-syn modulates key events in the pathogenesis of synucleinopathy such as: (i) variations in the fibrillar structure; (ii) different propagation properties; (iii) increase in cytotoxicity [17][53].
Indeed, a diversity of pathogenic properties of the misfolded conformations and accumulating aggregates of α-syn have been associated with: (i) mitochondrial dysfunction; (ii) endoplasmic reticulum stress; (iii) proteostasis dysregulation; (iv) synaptic impairment; (v) cell apoptosis; (vi) neuroinflammation; and (vii) neurodegeneration [18][19][20][21][22][23][11,54,55,56,57,58].
Notably, the pathological α-syn aggregates may spread from one neuron to another, causing Lewy pathology in the whole brain [24][59]. However, α-synuclein-positive inclusions have also been found in the cytoplasm of oligodendrocytes, an event that occurs in the α-synucleinopathy called multiple system atrophy. Specifically, Braak et. al. (2002) suggested that pathological forms of the α-syn act in a prion-like manner, trafficking between cells in a non-random way/form [24][59]. They hypothesized that PD pathology initiates in the peripheral nervous system, gaining access to the central nervous system (CNS) through retrograde transport via the olfactory tract and the vagal nerve [24][25][59,60]. It has been argued that the release and propagation mechanisms of α-syn between neuroanatomically connected regions can be through exosomes, classical exocytosis, trans-synaptic junctions, tunneling nanotubes, and direct penetration [26][61]. Last but not least, recent studies have suggested that α-syn misfolding and aggregation trigger microglial activation, leading to neuroinflammation and cellular metabolic stress, enhancing the aggregation and spreading of α-syn and affecting its prion-like transmissibility and pathogenicity (Braak’s hypothesis) [27][28][29][30][62,63,64,65].
3. The Misfolding of α-Synuclein and Its Association with Neuroinflammation in Parkinson’s Disease
A hypothesis claims that chronic neuroinflammation can lead to neuronal damage, neuronal circuitry disturbances, and ultimately, neurodegeneration in PD [31][32][20,66]. Hence, chronic neuroinflammation is relevant when considering the pathophysiological mechanisms involved in PD progression and proposing appropriate therapeutic approaches. The brain is an organ susceptible to external stimuli. However, internal stimuli can also alter the delicate homeostasis of the neuronal microenvironment maintained by microglia and astrocytes, considered the brain’s absorptive, excretory, and defense systems [33][67]. These cells display a Janus-like face because they help eliminate neurotoxins and pathogens, and conversely, they can also cause neuroinflammation, neurotoxicity, and neurodegeneration. Thus, neuroinflammation is a complex pathological condition where different cells and humoral factors converge to resolve the damage as a first intention and later they aggravate the disease in the long term. The cellular actors are activated microglia, reactive astrocytes, and infiltrated lymphocytes, whereas the humoral factors are a great variety of pro-inflammatory molecules. A resulting critical event from the flare-up between cells and humoral factors activities is the loss of BBB permeability that allows molecules to cross from one side to another of the brain [32][66].
In PD, microglia activation can arise from several factors or causes. The preference of activated microglia for brain areas enriched with pathological α-syn aggregates supports its close association with the neurodegeneration process in PD [34][35][68,69]. Multiple studies have shown that extracellular α-syn stimulates microglial cells to produce pro-inflammatory molecules such as interleukin (IL)-1β, IL-6, tumoral necrosis factor-alpha (TNF-α), cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), and reactive oxygen species (ROS) [36][37][38][39][40][70,71,72,73,74]. The combined neuroinflammation and oxidative stress can promote the neurodegenerative process and further aggravate it [41][42][75,76].
Furthermore, neuron-derived α-syn can stimulate astrocytes to produce and release pro-inflammatory cytokines and chemokines, which in turn can recruit activated microglial cells [43][12] and differentiate them to an M1 (pro-inflammatory) or M2 (anti-inflammatory) phenotype [44][77]. Therefore, pathological α-syn also behaves as a chemokine to concentrate activated microglia in the affected anatomical areas in PD [45][46][78,79].
The relevance of activated microglia in neuroinflammation and neurodegeneration relies on its ability to convert physiological astrocytes into neurotoxic reactive astrocytes classified as A1 phenotype. Recently, it has been established that IL-1α, TNF, and complement component 1q (C1q) released by activated microglia are sufficient and necessary to detonate the A1 phenotype (Figure 2a) [47][80]. Furthermore, evidence in vitro and in vivo shows that neurotoxic reactive astrocytes A1 can kill neurons through the secretion of neurotoxins not yet identified [47][48][80,81]. Thus, the neurotoxic role of reactive astrocytes A1 has severe implications for PD and other neurodegenerative diseases. It means that neurotoxic astrocytes lost their ability to promote neuronal survival, growth, synaptogenesis, and phagocytosis. Therefore, an effective therapy must also prevent the conversion of neurotoxic reactive astrocytes A1 and block their neurotoxic activity [47][48][80,81].
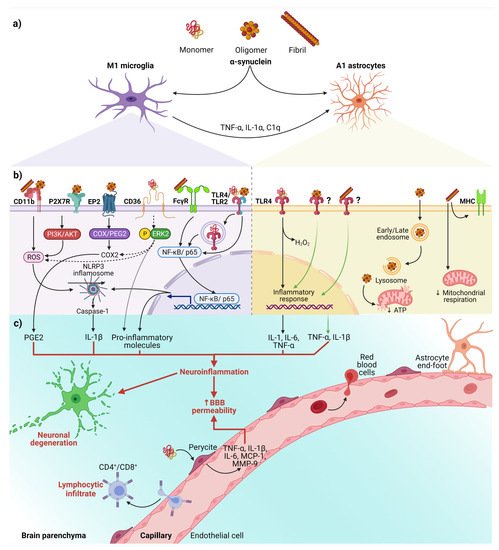
Figure 2. The interaction between α-syn and neuroinflammation in PD. (a) Activation of glial cells by pathological α-syn aggregates; (b) Signaling pathways in microglia and astrocytes triggered by interaction with the different aggregation patterns of α-syn; (c) Neuronal and BBB dysfunction triggered by the neuroinflammatory environment. Dash lines indicate the signaling pathway activated by CD36; Question marks and faded green lines indicate the proposed mechanism for oligomeric and fibrillar α-syn interaction with astrocytes, and thick red lines indicate the proinflammatory molecules that lead to dysfunction of the BBB and neural degeneration. Abbreviations: AKT, Protein kinase B; BBB, Blood-brain barrier; C1q, complement component 1q; CD, cluster of differentiation; COX, cyclooxygenase; EP2, E prostanoid receptor 2; ERK2, extracellular signal-regulated kinase 2; FcγR, the gamma chain subunit of Fc receptor; H2O2, hydrogen peroxide; IL, interleukin; MCP-1, Monocyte Chemoattractant Protein-1; MHC, Major Histocompatibility Complex; MMP-9, Matrix metallopeptidase 9; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NLR family pyrin domain containing 3; P2X7R, P2X7 receptor; PGE2, Prostaglandin E2; PI3K, phosphoinositide 3-kinase; ROS, reactive oxygen species; TLR, Toll-like receptors; TNF-α, tumor necrosis factor-alpha. Created with BioRender.com.
Besides, the interaction between α-syn with glial cells depends on α-syn aggregation state and the receptors responsible for its uptake (Figure 2b). These receptors expressed in glial cells, through which α-syn interact to trigger a neuroinflammatory environment (Table 1 and Figure 2b,c), play a critical role in early PD, considering that the α-syn aggregation process from soluble oligomers to insoluble inclusions occurs in the early phase of the disease [49][50][15,82].
Table 1.
Neuroinflammatory and neurotoxic effects triggered by pathological α-syn interaction with glial cells.
| α-Syn Aggregation Pattern | Glial Receptor/Mechanism | Signaling Pathway | Neuroinflammatory/Neurotoxic Effects | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fibrils | NLRP3 | a | α-syn acts as DAMP and activates the NLRP3 inflammasome. | Synthesis and release of IL-1β and cleaved caspase-1 that triggers pyroptosis *. | [49][51][52]] | [15,83 | [ | ,84 | 53] | ,85 | [54] | ,86 | [55 | ,87] |
| Monomers, oligomers, and fibrils | TLRs | a,b, | † | TLRs sense DAMPs, including α-syn, leading to nuclear translocation of NF-κB. | Release of pro-inflammatory cytokines (TNF-α and IL-6). Dual effect on the astrocyte: secretion of pro-inflammatory and/or neuroprotective factors. | [43][49][56][57][58][59][60][61] | [12,15,16,17,88,89,90,91] | |||||||
| Fibrils | FcγR | a | Internalization in phagosomes and nuclear translocation of NF-kB p65. | Clearance of α-syn, triggering the release of pro-inflammatory molecules and neurodegeneration. | [49][62][63] | [15,92,93] | ||||||||
| Oligomers and fibrils | CD11b | a | NOX2 activation through a RhoA-dependent pathway. |
NOX2 activation mediates the chemoattractant ability of α-syn. Induction of superoxide production. |
[49][46][64] | [15,79,94] | ||||||||
| Oligomers | EP2 | a | The cyclooxygenase/prostaglandin E2 (COX/PGE2) pathway. | Activation of PHOX NADPH and increase in prostaglandin E2 levels, leading to neuronal toxicity. | [49][65][66][67] | [15,95,96,97] | ||||||||
| Monomers | CD36 | a | Phosphorylation of ERK2, a downstream kinase activated by CD36 ligation. | Neuronal death through the release of TNF-α and ROS and up-regulation of COX2, NOX2, and iNOS. | [49][38] | [15,72] | ||||||||
| Oligomers | P2X7R | a | Activation of the PI3K/AKT pathway. | Increase of oxidative stress by p47phox translocation and PHOX activation. | [49][68] | [15,98] | ||||||||
| Fibrils | MHC | b | Changes in the expression of | HLA | genes encoding MHC class I and II proteins. | Impairment of ATP-generating mitochondrial respiration. | [69] | [99] | ||||||
| Oligomers | Endocytosis | b | Dysfunction in mitochondrial dynamics. | Neuronal death is mediated by cytokines release. | [70] | [100] |
Abbreviations: AKT, Protein kinase B; ATP, Adenosine triphosphate; CD, cluster of differentiation; COX, cyclooxygenase; DAMP, damage-associated molecular pattern; EP2, E prostanoid receptor 2; ERK2, extracellular signal-regulated kinase 2; FcγR, The gamma chain subunit of Fc receptor; HLA, human leukocyte antigen; IL, interleukin; iNOS, inducible nitric oxide synthase; MHC; major histocompatibility complex; NADPH, nicotine adenine dinucleotide phosphate; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NLR family pyrin domain containing 3; NOX2, NADPH oxidase 2; P2X7R, P2X7 receptor; PGE2, Prostaglandin E2; PHOX, phagocytic oxidase; PI3K, phosphoinositide 3-kinase; ROS, reactive oxygen species; TLR, Toll-like receptors; TNF-α, tumor necrosis factor-alpha. * An inflammatory type of cell death that causes excessive cell swelling, membrane rupture, and cytokines release. a Microglia and b astrocytes. † Oligomers for TLR-2; monomers, oligomers, and fibrils for TLR-4.
On the other hand, an in vitro study demonstrated that monomeric α-syn activates pericytes (critical cells in BBB regulation) to release pro-inflammatory molecules and matrix metalloproteinase-9 (MMP-9) [71][101]. These findings suggest that pericytes could exacerbate the neuroinflammatory environment and cause the BBB rupture in PD patients (Figure 2c).
Finally, the infiltrated peripheral immune cells, especially CD4+ and CD8+ T lymphocytes, extend and worsen the PD pathology (Figure 2c) [72][73][22,102]. This complication shows the critical role BBB breakdown plays in PD, thus becoming a point to control by new therapeutics. Moreover, infiltrated T lymphocytes, recognizing pathological α-syn aggregates as an antigen presented by microglial class II major histocompatibility complex (MHC-II), arise the immune response [74][103]. The increased infiltration of Th17 cells and reactive Th1 cells differentiated from CD4+ lymphocytes in PD brains proves the immune response’s involvement in this neuropathology [73][75][102,104]. Therefore, these antecedents support the immune response as another mechanism of neuronal death and promote the development of effective immunotherapies.
In summary, we propose the following sequential cellular events that lead to neuroinflammation in early PD (Figure 2). First, misfolded α-syn binds to microglial receptors causing differentiation of the microglia into the M1 phenotype. Then, M1 microglia release TNF-α, IL1α, and C1q inducing the conversion of astrocytes into neurotoxic reactive astrocytes A1. Both, the M1 microglia and A1 astrocytes release pro-inflammatory cytokines to open the BBB and chemokines to attract CD4+ and CD8+ cells, thus completing an immune response against misfolded α-syn. Moreover, the action of neurotrophic factors and other protective molecules released by astrocytes A2 is overpassed by the neuroinflammatory events. Altogether, the unknown molecules released by neurotoxic reactive astrocytes A1, the pro-inflammatory cytokines, and the cellular and humoral response of professional immune cells converge to kill the DA neurons in the early stages of PD. Therefore, new antiparkinsonian therapies should prevent α-syn binding to its glial receptors, block microglial M1 and astrocytic A1 activities, strengthen BBB permeability and avoid activation of the immune response. However, present therapeutic effects are insufficient for an integral PD therapy since the structural and functional restoring of the nigrostriatal dopaminergic system is not accomplished.