Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Jason Zhu and Version 2 by Jason Zhu.
microtubules and homologous recombination nucleoprotein filaments, where a network of intrinsically disordered tails exerts regulatory function in recruiting partner macromolecules, proteins or DNA and tuning the atomic level association.
- molecular modeling
- fuzzy complexes
- disorder and function
1. Functional Role of the Fuzzy Interface in the Cell
A growing body of reported observation on fuzzy interfaces depicts a continuum of association properties that range from quasi non-selective, liquid-like interactions to highly specific interactions, resulting from already mentioned folding-upon-binding mechanisms. Liquid-like association aims at ensuring proximity between the partner macromolecules and mostly involves electrostatic or polar interactions. Disordered proteins are a major component of membraneless cellular compartments, where they participate in liquid–liquid phase separation while avoiding aggregation, via the formation of dynamic, multivalent interactions [111,112][1][2]. Interestingly, high level of disorder in fuzzy interfaces are not necessarily associated to low affinity: in the complex between the human proteins histone H1 and its nuclear chaperone prothymosin-α, large opposite net charges have been shown to confer picomolar affinity to the association in spite of the absence of defined binding sites [113][3].
Researchers examine intermediate situations where disordered proteins or segments present ubiquitous motifs that can transiently associate to defined binding sites on folded protein partners. We more specifically address cases where both structurally organized and disordered regions coexist in the same protein. Typical examples are proteins that present disordered C- or N-terminal tails, largely represented among DNA-binding or DNA-processing proteins. The disordered tails in these proteins generally present a net charge. Positive tails can assist the efficiency of DNA search for specific sequences by proteins such as transcription factors. The tails can non-selectively bind to DNA and promote inter-segment cross talks in a “monkey-bar”-type mechanism [114,115][4][5]. When they bear a net negative charge, the tails can compete with DNA for binding sites [116,117][6][7] or they can bind one or more protein partners. For example, the tetrameric SSB protein that binds DNA single strands, an essential contributor of DNA replication, recombination, and repair in bacteria, functions as a recruitment platform where its four negatively charged C-terminal tails can simultaneously bind one or more proteins, thus favoring the transfer of bound DNA to these partner proteins [118,119][8][9]. Competition and recruitment mechanisms are also common in self-associating proteins that present disordered tails, such as tubulin or fibrinogen [120,121][10][11]. In those cases, the strongly charged tails actively contribute to the binding of partner subunits using fly-casting types of mechanisms, but do not participate in the protein–protein interface once the assembly is formed. In the case of tubulin associating into microtubules, the tails form molecular brushes around the microtubule lattice and participate in active or passive diffusion of proteins along the microtubule protomers [122][12].
The delicate balance between binding and unbinding provides the disordered terminal tails affinity tuning functions: the tails have been shown to modulate binding behaviors in response to changes in salt concentration or composition. The tail properties are also very sensitive to changes in the distribution of their charges resulting from post-translational modifications, as well as associated excluded volume modifications [121][11].
2. Interactions between the C-Terminal Tails of α,β-Tubulin Dimers and the Tubulin Core
Tubulin proteins exist in the cell as dimers of α- and β-tubulin, two closely related proteins whose sequences essentially differ at the level of their disordered C-terminal tails; both tails bear a net negative charge but they differ in length and amino-acid composition. α,β-tubulin dimers are the building blocks of microtubules (MT), the largest components of the cytoskeleton, that form highways for intracellular trafficking as well as separating chromosomes during meiosis. Modeling and NMR studies have shown that in tubulin dimers, both α - and β-tails can interact with the structurally organized region of the protein dimer (the core region) in spite of the core surface potential being mainly negative [95][13]. The tails are also known to contribute to the formation of microtubules by favoring the proper uptake of new tubulin dimers within the tubular architecture: alternative association forms of tubulin could be observed in the absence of tails [123][14]. It is therefore likely that the MT tubulin tails interact with free tubulin dimers during the assembly process, thus orienting the dimers toward the desired binding geometry. This association however needs to be transient, since a large fraction of the tails (notably the longer β-tails) are released during the process and become free to interact with microtubule-binding proteins (MAPs) [121][11]. Similar process has been observed by AFM when fibrin proteins assemble into fibrinogen [120][10], while the C-terminal tails of RecA proteins have been shown to be involved in their association process into filaments [124][15]. These observations indicate that the ability of the disordered protein tails to bind the protein core surface but also to unbind from it is key to their function.
How exactly the tails influence auto-assembly remains to be established. Theoretical simulation of the tubulin tails binding to their associated dimeric protein cores enabled to gain insights on this question [95][13]. Notably, while the surface spanned by the tails during atomic molecular dynamics simulations was found compatible with ensemble observations obtained by AFM (radius of gyration), the simulation enabled proposing a finer characterization of the spatial and temporal distribution of the tails, based on specifically developed metrics using the position of the tail center of mass, together with time analysis of the contacts between tails and protein cores. This analysis revealed the presence of a handful of specific tail-binding spots, or anchors, distributed on the tubulin surface and presenting reduced surface areas. The tails develop versatile interactions with these binding spots, mostly based on electrostatic complementarity [95,121][13][11]. Interestingly, negatively charged amino-acid patches distributed along the whole β-tail (see Figure 1) can individually bind separate binding spots, and that adjacent negative patches can slide within a given anchor and exchange their binding interactions. Binding different sites on the core surface does not seem to be cooperative but rather self-exclusive, one reason being that several negative patches on the tail may not be able to simultaneously access spatially separated anchors. Another factor arises from the electrostatic potential around the tubulin dimer. Indeed, the electrostatic potential partitions the space available to the tails into electronegative regions, that are strongly repulsive for the most part of the tail length, and electropositive funnels that strongly attract the negative tail patches. This situation creates tension and frustration in the bound tails, part of which needs to reside in an unfavorable, repulsive region to allow contacts to form on the tubulin surface [95][13]. Frustration, a tradeoff between conflicting forces within their interatomic contact network environment [125][16], has been identified as a critical property of IDPs or IDRs binding to their protein targets [13][17]. Because of unsolved conflicts at such interfaces, added to the multiplicity of binding sites, the disordered regions are prone to switching to alternate binding geometries. The concept of frustration extends to long-range interactions such as the response of protein tail conformations to the potential energy created by the protein core, coupled to the physical attachment between the tail and the protein core. Long range frustration may constitute a powerful driving force to facilitate the tail unbinding from its core protein. It is also easily tunable via changes in the salt concentration or modification of the charge distribution in the tail via post-translational modifications (PTM). Indeed, recent work from Bigman and Levy showed that PTMs tune the binding ability of the tails to the MT, a function that is also partly linked to their exclusion volume properties [121][11]. Recent simulations of the hepatitis B virus (HBV) Core protein, that exhibits a 33-residue long, positively charged and intrinsically disordered C-terminal tail, suggests the existence of long range frustration in the binding of the negatively charged extremity of the tail to the positively charged extremity of the HBV capsid spike, with very sparse interactions between the rest of the tail and the external surface of the spikes [126][18].
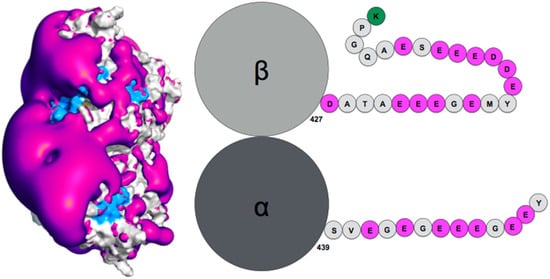
Figure 1. (Left) Negative surface electrostatic potential (-1 kT, magenta) of the αI/βIII isotype tubulin body without tails. The anchor residues involved in interactions with the disordered tail during molecular dynamics simulations are shown in blue; the representation is based on data published in (Laurin et al., Biochemistry 2017, 56, 1746); (right) schematic sequence of the αI/βIII isotype of tubulin, the acidic amino acids are highlighted in magenta and the basic terminal residue in green.
3. Role of the RecA Protein C-Terminal Tails in Homologous Recombination
Homologous recombination permits the faithful repair of DNA double strand breaks in the genome, by recruiting intact genomic DNA (dsDNA) with sequence similar to the damaged DNA and using that DNA to restore the lost sequence continuity. To this aim, the dsDNA complementary strand is captured by a single strand (ssDNA) from the damaged DNA, in a process called strand exchange that occurs within filaments of recombinase proteins (RecA in bacteria) [127][19]. Alike many proteins that process DNA, E. coli RecA proteins present a disordered terminal tail, here a 25-amino acids, negatively charged C-terminal tail with seven acidic amino-acids. The C-terminal tail was shown to participate in the regulation of various stages of the recombination process: the filament self-assembly, the intake of the dsDNA into the filament, and the yield of strand exchange. While all those stages can take place in the absence of the tail or with partly deleted tails, the tail has been shown to mediate the response of the process to changes in pH or in magnesium concentration [124,128,129][15][20][21]. Specifically, full-length tails slow the RecA self-association process but promote the formation of longer and more stable filaments on ssDNA [124][15]. During the search, the dsDNA intake is also slowed in the presence of the tail, but this effect is reduced by adding 2mM free Mg2+ ions. This observation has been related to the fact that the searched dsDNA non-specifically binds to the filament gateway [130][22], a region that crosses the filament groove and involves basic amino-acids from the C-terminal domains. The acidic C-terminal tails may restrict the access of the dsDNA to the gateway via electrostatic repulsion or physical steric hindrance and the added magnesium ions may reduce the electrostatic repulsion and possibly induce the formation of secondary structures in the tails, which would confine the tails in a smaller volume.
How the disordered tail influences the strand exchange process is more puzzling. In the presence of the disordered tails, addition of 5 mM magnesium ions maximizes the formation of the strand exchange product; the magnesium concentration has no effect if the tail has been deleted, indicating that the tail is fully involved in the process. It has been proposed that in condition of low magnesium concentration, the tail may compete with the incorporated dsDNA for binding to the filament secondary binding site (site II) [128,129][20][21]. In that hypothesis, the tail may stimulate dsDNA binding to site II by disengaging from that site following changes in magnesium concentration [128][20]; alternatively, the tail may assist unbinding of non-homologous incorporated dsDNA, thus accelerating the search process [124][15]. However, site II is buried in the filament interior whereas the tail extremities are situated at the periphery of the filament [131,132][23][24] (Figure 2). In order for its acidic residues to reach site II in the filament interior, the tail would need to adopt a stretched conformation along the C-terminal domain toward the filament interior. The first exploration of the tail structural dynamics by molecular dynamics simulations did not show such behavior [129][21]. Instead, all seven tails of a simulated filament, made of seven RecA monomers bound to a 21-nt ssDNA, remained at the exterior of the filament during the course of two 200 ns simulation with no added or 2 mM magnesium ions. The tails partly formed helical folds and partly lied on the external surface of the filament, sometimes spanning over two consecutive monomers, but they did not penetrate into the filament interior. Recently, study further explored the tail dynamics by taking into account the perturbation induced in the filament structure by the hydrolysis of ATP molecules situated at the interface between monomers. Indeed, experimental observation of the influence of the tails on the strand exchange process was performed in conditions of ATP hydrolysis, using ATP regeneration system. Recent modeling studies indicate that the response of the filament to ATP hydrolysis may involve important modifications in the spatial partitioning of the filament groove, which may modify the tail accessibility to the filament interior. Researchers used published model of a 2-turn (12 monomers) filament [133][25] where the central RecA-RecA interface was modified for an ADP interface (the RecA-RecA binding geometries differ whether the cofactor is ATP or ADP) as a starting point for two 100-ns molecular dynamics simulations, one with no added magnesium ions and one with 5 mM magnesium concentration. Interestingly during the simulation with 5 mM magnesium, the tail associated to the central monomer with modified interface spontaneously penetrated in the filament interior and reached the secondary DNA binding site, showing that this proposed behavior is indeed topologically and energetically possible within filament architectures associated to ATP hydrolysis (Figure 2). These preliminary simulations need to be replicated and call for further investigation in order to draw any reliable conclusions on the effects of the magnesium concentration; notably, force fields adapted not only to different levels of structural disorder but that also correctly capture magnesium ion interactions need to be tested in order to confirm the reported observations. Magnesium ions can individually mediate interactions between negative charges but can also as an ensemble contribute to weaken salt-bridge interactions, therefore contributing to order-disorder transitions. Molecular dynamics is a tool of choice for disentangling individual from ensemble effects of the magnesium ions, provided that the interactions are correctly accounted for. Present MD observations are too preliminary to conclude about the exact role of the magnesium, nevertheless they point to topological and steric factors as additional factors for the tails to exert their control function.
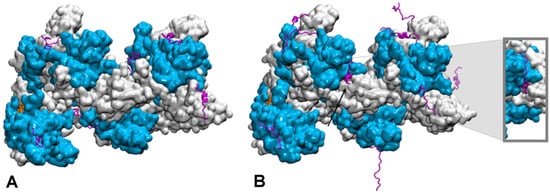
Figure 2. Conformational dynamics of the C-terminal tails of a two-turn, twelve monomer RecA-ssDNA filament with modified central interface, after 100 ns of molecular dynamics simulation. Successive RecA proteins are alternatively colored cyan and white. The tails (magenta, cartoon representation) explore different regions of the conformational space in terms of folding—partial α-helical folds or extended conformation—and binding to the protein core surface. (A) Simulation with no added salt; the tails mostly bind the core protein surface; (B) simulation with 5 mM Mg2+; some tails remain far from the surface, the tail from the central monomer (black arrow) penetrates inside the filament and reaches the filament site II, within 8 Å of the basic residue cluster of the neighboring monomer. The insert shows a view of the penetrating tail after 30° rotation around the filament axis. Simulations conditions are described in (Kim et al., Nucleic Acids Res. 2018, 46, 2548).
References
- Cuevas-Velazquez, C.L.; Dinneny, J.R. Organization out of disorder: Liquid-liquid phase separation in plants. Curr. Opin. Plant Biol. 2018, 45, 68–74.
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 2018, 18, e1700193.
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66.
- Khazanov, N.; Levy, Y. Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J. Mol. Biol. 2011, 408, 335–355.
- Vuzman, D.; Levy, Y. Intrinsically disordered regions as affinity tuners in protein-DNA interactions. Mol. Biosyst. 2012, 8, 47–57.
- Shishmarev, D.; Wang, Y.; Mason, C.E.; Su, X.C.; Oakley, A.J.; Graham, B.; Huber, T.; Dixon, N.E.; Otting, G. Intramolecular binding mode of the C-terminus of Escherichia coli single-stranded DNA binding protein determined by nuclear magnetic resonance spectroscopy. Nucleic Acids Res. 2014, 42, 2750–2757.
- Mondal, A.; Bhattacherjee, A. Mechanism of Dynamic Binding of Replication Protein A to ssDNA. J. Chem. Inf. Model. 2020, 60, 5057–5069.
- Shereda, R.D.; Bernstein, D.A.; Keck, J.L. A central role for SSB in Escherichia coli RecQ DNA helicase function. J. Biol. Chem. 2007, 282, 19247–19258.
- Marceau, A.H.; Bahng, S.; Massoni, S.C.; George, N.P.; Sandler, S.J.; Marians, K.J.; Keck, J.L. Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J. 2011, 30, 4236–4247.
- Protopopova, A.D.; Litvinov, R.I.; Galanakis, D.K.; Nagaswami, C.; Barinov, N.A.; Mukhitov, A.R.; Klinov, D.V.; Weisel, J.W. Morphometric characterization of fibrinogen’s alphaC regions and their role in fibrin self-assembly and molecular organization. Nanoscale 2017, 9, 13707–13716.
- Bigman, L.S.; Levy, Y. Modulating Microtubules: A Molecular Perspective on the Effects of Tail Modifications. J. Mol. Biol. 2021, 433, 166988.
- Bigman, L.S.; Levy, Y. Tubulin tails and their modifications regulate protein diffusion on microtubules. Proc. Natl. Acad. Sci. USA 2020, 117, 8876–8883.
- Laurin, Y.; Eyer, J.; Robert, C.H.; Prevost, C.; Sacquin-Mora, S. Mobility and Core-Protein Binding Patterns of Disordered C-Terminal Tails in beta-Tubulin Isotypes. Biochemistry 2017, 56, 1746–1756.
- Bhattacharyya, B.; Sackett, D.L.; Wolff, J. Tubulin, hybrid dimers, and tubulin S. Stepwise charge reduction and polymerization. J. Biol. Chem. 1985, 260, 10208–10216.
- Fan, H.F.; Su, S. The regulation mechanism of the C-terminus of RecA proteins during DNA strand-exchange process. Biophys. J. 2021, 120, 3166–3179.
- Ferreiro, D.U.; Komives, E.A.; Wolynes, P.G. Frustration in biomolecules. Q. Rev. Biophys. 2014, 47, 285–363.
- Freiberger, M.I.; Wolynes, P.G.; Ferreiro, D.U.; Fuxreiter, M. Frustration in Fuzzy Protein Complexes Leads to Interaction Versatility. J. Phys. Chem. B 2021, 125, 2513–2520.
- Carvaillo, J.-C. From Assembly Unit to Capsid: In Silico Application to Norovirus and Hepatitis B Virus; Université Paris-Saclay: Gif-sur-Yvette, France, 2021.
- Bell, J.C.; Kowalczykowski, S.C. RecA: Regulation and Mechanism of a Molecular Search Engine. Trends Biochem. Sci. 2016, 41, 491–507.
- Lusetti, S.L.; Shaw, J.J.; Cox, M.M. Magnesium ion-dependent activation of the RecA protein involves the C terminus. J. Biol. Chem. 2003, 278, 16381–16388.
- Kim, R.; Kanamaru, S.; Mikawa, T.; Prevost, C.; Ishii, K.; Ito, K.; Uchiyama, S.; Oda, M.; Iwasaki, H.; Kim, S.K.; et al. RecA requires two molecules of Mg2+ ions for its optimal strand exchange activity in vitro. Nucleic Acids Res. 2018, 46, 2548–2559.
- Kurumizaka, H.; Aihara, H.; Ikawa, S.; Kashima, T.; Bazemore, L.R.; Kawasaki, K.; Sarai, A.; Radding, C.M.; Shibata, T. A possible role of the C-terminal domain of the RecA protein. A gateway model for double-stranded DNA binding. J. Biol. Chem. 1996, 271, 33515–33524.
- Chen, Z.; Yang, H.; Pavletich, N.P. Mechanism of homologous recombination from the RecA–ssDNA/dsDNA structures. Nature 2008, 453, 489–494.
- Yang, D.; Boyer, B.; Prevost, C.; Danilowicz, C.; Prentiss, M. Integrating multi-scale data on homologous recombination into a new recognition mechanism based on simulations of the RecA-ssDNA/dsDNA structure. Nucleic Acids Res. 2015, 43, 10251–10263.
- Boyer, B.; Danilowicz, C.; Prentiss, M.; Prevost, C. Weaving DNA strands: Structural insight on ATP hydrolysis in RecA-induced homologous recombination. Nucleic Acids Res. 2019, 47, 7798–7808.
More