Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Consolato Sergi and Version 2 by Catherine Yang.
Sarcoidosis is a non-necrotizing granulomatous inflammatory syndrome with multisystemic manifestations.
- Paediatric sarcoidosis
- high-risk sarcoidosis
- pulmonology
- granulomatous disease
- chronic inflammation
- children
- lung
- respiratory system
- pathology
- consulting
1. Introduction
Sarcoidosis is a multisystemic syndrome with a highly variable clinical course and diverse disease manifestations [1]. The incidence of sarcoidosis in adults may be biphasic [2]. Historically, it was thought to commonly affect young adults 30–50 years of age, but recent studies have reported that more than half of incident diagnoses are made in patients over 55 years of age [2][3][2,3]. Erdal and others suggest that the rates of sarcoidosis are increasing [4]. Approximately 25% of affected individuals with the disease develop chronic and progressive disease, which contributes to increased disease burden [5][6][5,6]. The mortality rate also appears to be rising [7]. There is no single diagnostic test for sarcoidosis. Instead, the diagnosis relies on specific pathological and radiographic features in the appropriate clinical settings. The disease is characterized by pathologic findings of non-necrotizing granulomas in one or more involved organ systems after alternative diagnoses, in particular, infective etiologies, have been entertained [8].
Sarcoidosis is an ever-evolving process. The clinical phenotypes range from single-organ, self-limited, asymptomatic disease to multi-organ involvement with high-risk manifestations [9]. Hilar lymphadenopathy and pulmonary interstitial infiltrations are the most common manifestations [10]. The term “high-risk sarcoidosis” was introduced at the National Heart Lung and Blood Institute Sarcoidosis Workshop 2017 [1] to denote several manifestations of sarcoidosis that are associated with impaired quality of life and relatively high risk of death [9]. These include treatment-resistant pulmonary sarcoidosis, cardiac sarcoidosis, neurosarcoidosis, and/or multi-organ involvement. The high-risk manifestations and multi-organ involvements are often missed until late in the disease course [9].
2. High-Risk Sarcoidosis
Approximately 25% of affected individuals with sarcoidosis develop chronic or progressive disease [6]. Manifestations of sarcoidosis associated with poor prognosis and a relatively high risk of death include treatment-resistant pulmonary-, cardiac-, neuro-, and multiorgan sarcoidosis [9]. Pulmonary disease is the most common cause of chronic disease and death in adults [9]. As in adults, older children usually present with hilar lymphadenopathy, and up to 50% present with interstitial lung disease and multiorgan involvement, while progression to chronic diseases occurs in 12% of these children [2][8][11][2,8,13]. Korsten et al. defined pulmonary sarcoidosis refractory to treatment as progressive disease and significant impairment of life despite glucocorticoid therapy for at least three months and the need for additional anti-sarcoid drugs [12][32]. These patients develop progressive pulmonary fibrosis and associated complications, including pulmonary hypertension and infections, and are seen in more than 10% of patients at specialized centers [12][13][32,33]. In infants and children younger than five years, the typical pulmonary diseases are usually not seen [8]. In a retrospective study of 41 pediatric patients, which were followed for 18 months on disease presentation, management, and clinical outcome, those patients with pulmonary sarcoidosis diagnosed before 10 years old were more likely to recover and presented with fewer relapses compared with the patients diagnosed after 10 years old [14][34].
Recent progress in cell-mediated immunity and granuloma formation has advanced our understanding of the development of sarcoidosis [8][15][8,35]. Macrophages bearing an increased expression of major histocompatibility (MHC) class II molecules, different subsets of T-lymphocytes, and other immune effector cells such as mast cells and natural killer cells may be at play [8][15][8,35]. In sarcoidosis involving the lung, before the formation of granulomas, early lesions consist of alveolitis with a high proportion of activated CD4+ T-cells [16][36]. T-cells play a role in the amplification of the local cellular immune response and are responsible for the secretion of cytokines, including tumor necrosis factor (TNF), which favors granulomatous response at the sites of disease activity. The diagnostic approach to pulmonary sarcoidosis has been revolutionized in the past decade by the use of endobronchial ultrasound (EBUS) real-time guided transbronchial sampling of intrathoracic lymph nodes and lung biopsies [6]. Cytopathological assessments by fine-needle aspiration provide fair diagnostic yield and excellent patient safety profile in children [17][18][37,38]. Combining EBUS and transbronchial needle aspiration (TBNA) lung biopsy and cytopathologic study may increase the diagnostic sensitivity to close to 100% [19][20][39,40]. On bronchoalveolar lavage (BAL) fluid with lymphocytosis (15%) and increased ratio of CD4 to CD8 (>3.5:1), the specificity for the diagnosis of sarcoidosis approaches 95% [16][21][36,41].
Cardiac sarcoidosis (CS) is a rare but potentially fatal condition [22][42]. Clinically recognizable cardiac involvement occurs in 5% of adult patients with sarcoidosis [23][43], and granulomatous inflammation of the heart was recognized in up to 25% in an autopsy series [11][24][13,44]. Rare cases of pediatric cardiac sarcoidosis had been reported [8]. Patients with CS may present with a wide variety of signs and symptoms, ranging from asymptomatic ECG abnormalities to sudden death [22][42]. Congestive heart failure and conduction abnormalities are the two most common clinical manifestations in CS [22][42], with one case presenting with pericardial effusion [2][8][2,8]. Many patients with pulmonary/systemic sarcoidosis have asymptomatic cardiac involvement [23][43]. A high index of suspicion and early diagnosis are crucial when immunosuppression therapy may result in a reduced mortality rate [8]. Various imaging modalities including echocardiography and/or CMR imaging may be utilized [23][43].
Neurosarcoidosis occurs in fewer than 5% of adults with systemic sarcoid with isolated neurosarcoidosis being rarer [25][45], and to our knowledge, 53 cases of pediatric neurosarcoidosis have been reported in the English literature [26][27][14,46]. In these children, the most common manifestations include headache, seizures, cranial neuropathy, optic neuritis, and hypothalamic and/or pituitary dysfunction [26][27][14,46]. Compared to their adult counterparts, prepubertal children with neurosarcoidosis are more likely to present with seizures and a space-occupying lesion but less likely with cranial nerve palsies. These children do however tend to evolve to an adult pattern of presentation during puberty [26][14]. Single cases of aqueductal stenosis [11][13] and obstructive hydrocephalus [28][47] have also been reported. The most reliable method for diagnosing neurosarcoidosis is via biopsy of the lesion in the central nervous system revealing non-caseating granuloma [27][46]. Microscopically, granulomatous inflammation is typically found in the meninges, ventricles (including choroid plexus), and adjacent brain or spinal cord parenchyma. Inflammatory infiltrate may involve optic nerve and chiasm, cranial nerves such as a facial, auditory, and vestibulocochlear nerve. In cases where biopsy is not possible, neuroimaging becomes an important diagnostic modality. The most common neuroimaging finding is said to be a leptomeningeal enhancement on T1-weighted MRI with contrast administration [29][48]. Enhancement may also be seen in the basilar region, leading to abnormalities in the hypothalamus, optic chiasm, and pituitary region [27][46]. As neurosarcoidosis is a diagnosis of exclusion, differential diagnoses that need to be considered and ruled out include tuberculosis, fungus, Wegener’s granulomatosis, and hypertrophic meningitis, as these may also present with leptomeningeal enhancement and granulomas. To the best of our knowledge, cases of pediatric spinal neurosarcoidosis have not been reported in the English literature.
3. Acute Sarcoidosis
In contrast to the chronic progressive disease with organ dysfunction in high-risk sarcoidosis, two acute sarcoidosis syndromes are of relatively benign clinical outcome. Lofgren syndrome is characterized by fever, bilateral hilar lymphadenopathy, erythema nodosum, and arthralgia, typically in young women, primarily Caucasian. Prognosis is excellent with complete resolution of the disease in 90% of the patients within two years [8][30][31][8,49,50]. Hereford syndrome or uveoparotid fever, presenting with a combination of fever, parotidomegaly, anterior uveitis, and cranial nerve palsy is usually seen in adults but has been reported in children [8][30][8,49]. The disease is normally self-limiting with a cure achieved between 1–3 years [8][32][8,51]. The acute sarcoidosis syndromes may be confidently diagnosed on clinical grounds without surgical biopsy confirmation in adults [8][30][33][8,49,52]. However, the clinical manifestations in children may be variable necessitating biopsy histopathology confirmation [8][34][30][35][8,15,49,53].
Pathology images of the spectrum of pediatric sarcoidosis involving different organs (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6 and Figure 7).
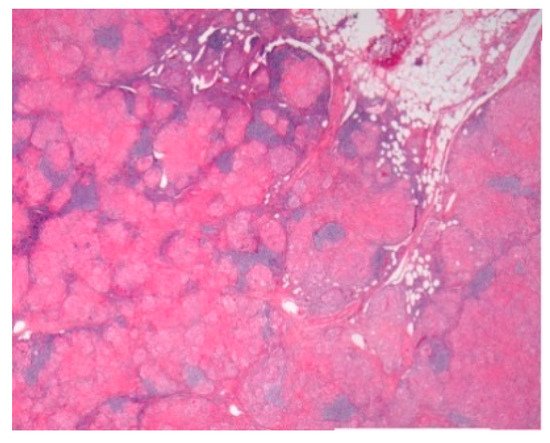
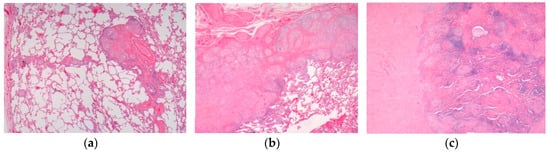
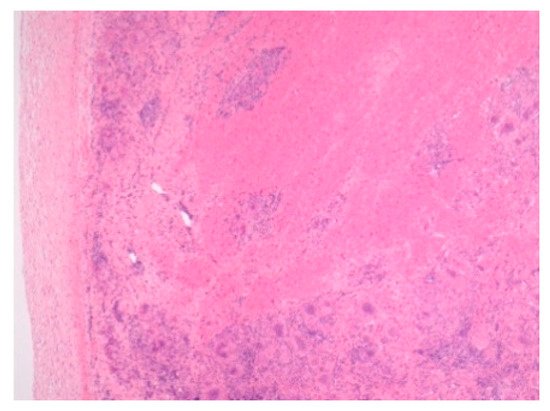
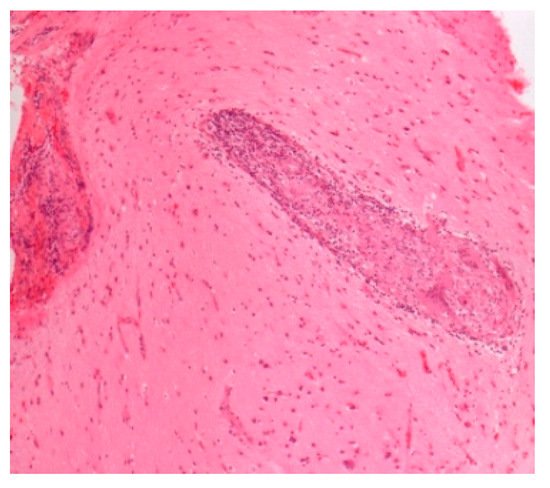
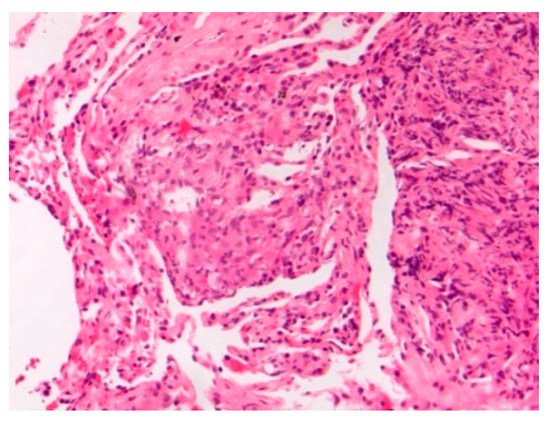
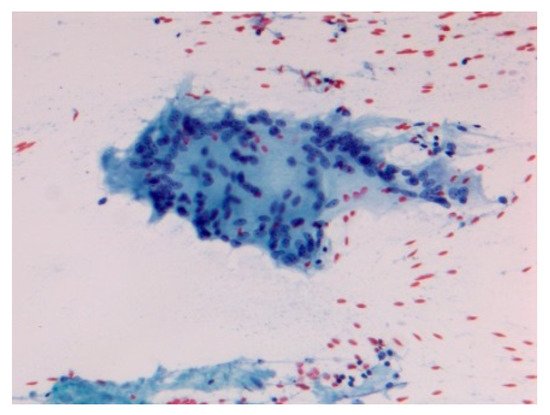
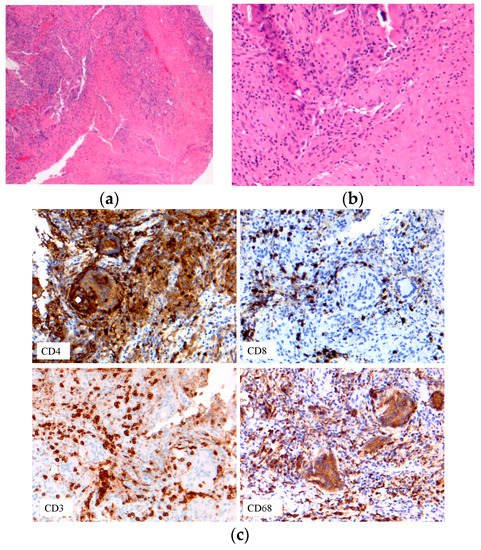
Figure 1.
Mediastinal nodal sarcoidosis. H&E Magnification 20×.
Figure 2.
Pulmonary sarcoidosis. (
a
) Pulmonary sarcoidosis. (
b
) Pulmonary nodular sarcoidosis. (
c
) Pulmonary fibrotic sarcoidosis. H&E. Magnifications 20×, 50×.
Figure 3.
Cardiac sarcoidosis. H&E Magnification 50×.
Figure 4.
Neurosarcoidosis. H&E. Magnification 100×.
Figure 5.
Lung sarcoidosis young E-cig vaper. H&E. Magnification 200×.
Figure 6.
Acute sarcoidosis salivary gland. Cytopathology Pap stain 200×.
Figure 7. Immune cell phenotypes: T-cells: CD3, CD4, CD8; macrophage CD68. (a) Early onset sarcoidosis (EOS) skin sarcoidosis; (b–c) EOS skin sarcoidosis.
Recent case reports of sarcoidosis developing after antigen exposure to inhalation or in immunotherapy are of interest. The popularity of electronic cigarettes (E-cig) has recently been increasing among adolescents and young adults [36][37][38][54,55,56]. E-cig usage or “vaping” contains E-liquid carrier components, flavorings, and marijuana products with cannabidiol (CBD) formulations [39][40][41][42][57,58,59,60]. In anecdotal stories, the use of medical marijuana, in particular vaping with CBD oil may be linked to an improvement of refractory pulmonary sarcoidosis [43][61]. In E-cig and marijuana users, sarcoid granulomas are found in multiple organs in case of reports, raising the clinical suspicion of metastatic malignancy [44][45][62,63]. The development of sarcoid granulomas may be related to vaped marijuana product tetrahydrocannabinol (THC), E-cig flavorings, or contaminants. An association of vaped THC has been reportedly seen in close to 500 cases of severe pulmonary disease with six confirmed deaths [38][56]. As tumor necrosis factor-alpha (TNF-α) plays an important role in both formation and maintenance of sarcoid granulomas, anti-TNF-α may provide a therapeutic option to patients with sarcoidosis [46][64]. However, many anti-TNF-associated cases of sarcoidosis have also been reported [47][65], negating its therapeutic usage. Pulmonary sarcoid granulomas are found in patients undergoing interferon-alpha therapy [48][66]. The use of immune checkpoint inhibitors by enhancing anti-tumor immunity has revolutionized cancer therapy recently, and their use for cancer immunotherapy in pediatric patients may have potential benefits [49][50][67,68]. However, pulmonary sarcoidosis and the exacerbation of sarcoidosis leading to central nervous system involvement after immune checkpoint inhibitor therapy have been reported [51][52][25,69].