Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Anja Bräuer and Version 2 by Catherine Yang.
Niemann–Pick disease type C1 (NPC1), which is a rare lipid-storage disorder. Lipids, in particular phospholipids, are a major component of the cell membrane and play important roles in cellular functions, such as extracellular receptor signaling, intracellular second messengers and cellular pressure regulation.
- neurodegeneration
- lipid storage disorder
1. Introduction
The rare Niemann–Pick disease type C1 (NPC1) is an autosomal–recessive, lipid-storage disorder characterized by neonatal jaundice, hepatosplenomegaly, and progressive neurodegeneration [1][2][3][1,2,3]. The mutation responsible for approximately 95% of these cases has been mapped to a gene on chromosome 18q11 designated NPC1 [4]. The progressive neurodegeneration induces ataxia, dystonia, and impairment of intellectual function [2][5][6][2,5,6]. The NPC1 protein is involved in intracellular lipid trafficking [7][8][7,8]. The defect caused by mutations in the NPC1 gene induces accumulation of unesterified cholesterol, glycosphingolipids, and other fatty acids in the endosomal/lysosomal system [9]. This impaired lipid transport leads particularly to an extensive loss of Purkinje cells in the cerebellum and degeneration of other central nervous compartments [10][11][12][13][10,11,12,13]. Although all NPC1 cells show cholesterol and glycosphingolipid accumulation, the major clinical impact is in the liver and brain [14]. Many affected individuals have liver disease at birth. Most begin to show symptoms of neurodegeneration as young children, with learning difficulties and motor coordination problems being paramount. These individuals typically die in their teen years. There is also an infantile form of the disease; these infants show hepatosplenomegaly, fail to thrive, and die within 2–3 years. Other individuals with a milder form of the disease enjoy a normal childhood and are diagnosed as adults, with early dementia as the predominant symptom. NPC was little studied in the past because of its rarity; NPC1 is diagnosed in one in every 92,000–150,000 births, though recent genome and exome analysis including late appearing phenotypes predicts an increased incidence of one in 20,000–39,000 births [15]. However, biomedical scientists took more notice when the NPC1 gene was shown to encode a large membrane protein with features shared by several key regulators of cholesterol homeostasis. Identification of the NPC2 protein by Lobel′s laboratory in 2000 [1] revealed a small soluble glycoprotein that likely partners with NPC1 in transporting lipids.
2. Lipid Trafficking and NPC1 (Niemann–Pick Disease Type C1)
2.1. Cholesterol Transport
Cholesterol homeostasis is essential for the functional integrity of the cell [3]. Nearly all cells in the body, including neurons of the central nervous system (CNS), take up cholesteryl ester and/or unesterified cholesterol carried in various lipoproteins from the surrounding pericellular fluid by receptor-mediated and bulk-phase endocytosis [16][17][16,17]. Both the cellular content and distribution of cholesterol within the cell are highly dynamic and tightly regulated through de novo synthesis of cholesterol by the endoplasmic reticulum (ER) [4], and by uptake of cholesterol ester-rich lipoprotein particles circulating in the serum by the low-density lipoprotein (LDL) receptor pathway [9]. The main sorting station for cholesterol within the cell is the late endosome (LE), an intermediate stage in the endosomal–lysosomal trafficking pathway. Two LE proteins, NPC1 and NPC2, appear to be key players that initiate the sorting process [18][19][20][21][18,19,20,21].
2.2. NPC Protein Function
2.2.1. NPC1
NPC1 is a large glycoprotein with 13 transmembrane-spanning domains that is found in LE [22]. It contains a five-transmembrane domain, called the ‘sterol-sensing domain’ that it is found in multiple other proteins hypothesized to sense the cholesterol content of their surroundings. A large hydrophilic N-terminal domain and two hydrophilic loops extend into the endosome lumen, but functions and/or binding partners are unknown. In the steady state, most NPC1 protein is found in LE, but the protein is present in tubules and vesicles that bud off from endosomes, traffic across the cell and then return [23]. The physiological importance of the NPC1 protein is emphasized by its conservation (yeast, insects, worms, and mammals all have NPC1), although these organisms diverge considerably in their need for, and handling of, sterols.
2.2.2. NPC2
The NPC2 gene encodes a protein with the reassuring features of a bona fide lipid-transport protein. NPC2 is a soluble glycoprotein that is delivered to lysosomes by virtue of its mannose phosphate moiety [1]. It is also secreted and found in epididymal fluid, bile, and milk. The secreted protein was purified in apo- and sterol-bound forms [24]. Apo-NPC2 was found to have an incipient ligand-binding pocket, which expands to accommodate cholesterol (Figure 1). NPC2 was shown, in in vitro assays, to rapidly transport cholesterol from donor to acceptor membranes via a collisional mechanism [25]. As might be expected for a lysosomal protein, transfer activity was greater in an acidic environment and was enhanced by the presence of the late-endosome/lysosome (LE/LY) -specific lipid lysobisphosphatidic acid.
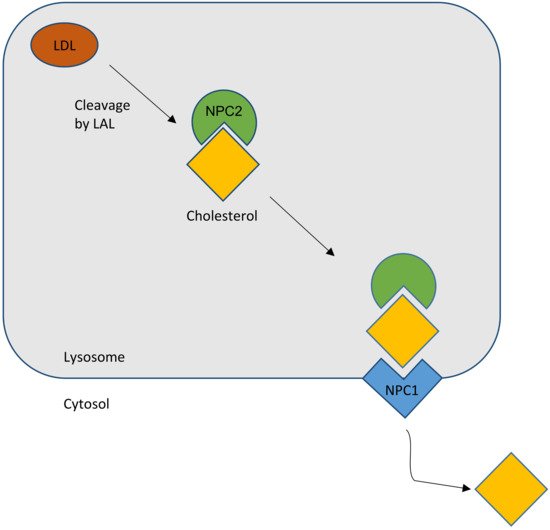
Figure 1. Schematic outline of the normal cholesterol trafficking via NPC2 and NPC1 interaction. LAL, lysosomal acid lipase (courtesy of René Thiemer, modified after [26]).
In summary, the most favored hypothesis is that, as a lipid cargo is brought to the LE/LY, the lipids are digested into their constituent molecules. NPC2 facilitates the transfer of cholesterol, and perhaps other lipids, to the delimiting membrane of the organelle. NPC1 senses the rising membrane cholesterol content and signals for the membrane to bud, carrying cargo to destinations throughout the cell (Figure 1).
2.3. Diagnostic Tools
The distinct heterogeneity of this disease makes it difficult to diagnose. There are several options to diagnose NPC: skin and liver biopsy for filipin staining of cultured fibroblasts; electron microscopic analysis of vacuolation or hepatocytes containing “myelin figures” (Figure 2) [27][28][29][27,28,29]; molecular genetic analysis with direct sequencing of NPC1 and NPC2 gene mutations [28]; bone marrow aspiration for the detection of foamy histiocytes [30][31][30,31]; and use of cholesterol esterification assays and oxysterol assay-based screening to measure the increase of cholestane-3β,5α,6β-triol (cholesterol oxidation product, “triol”) [32][33][34][35][32,33,34,35]. A possible non-chemical biomarker and treatment control may consist in olfactory testing, since olfactory deficits may mirror the progress of the disease [36]. However, as yet there are no human data available [27].
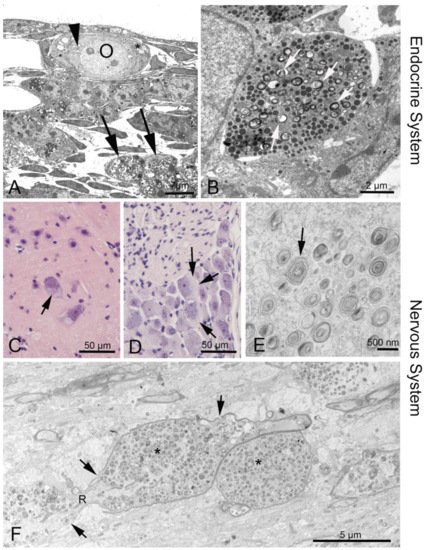
Figure 2. Phenotypes of NPC1 in the endocrine and nervous system. (A) Ovary of an Npc1−/− mouse. The oocyte (O) contains enlarged endoplasmic reticulum (ER) with myelin-like deposits (arrowhead), as does the surrounding follicular epithelial cell (asterisk). Large accumulations are seen in a macrophage (arrows). (B) Neuroendocrine cell in the anterior pituitary. Arrows point at myelin-like inclusions between secretory vesicles. (C) Some alpha motor neurons in the anterior horn of the spinal cord are filled with light material replacing the darker perinuclear Nissl substance of the endoplasmic reticulum. (D) Similar damage is seen in dorsal root ganglion cells (arrows). (F) Corpus callosum: A longitudinal nerve fiber is enlarged and congested by autophagosome content (asterisks) that interrupts the continuity of neurofilaments and neurotubuli. The myelin sheath (arrows) has also thinned and disintegrated. R, node of Ranvier.
2.4. Therapies
So far, there is no causal therapy of NPC1, though the iminosugar miglustat (Zavesca®) is the only approved drug in Europe used for supporting and symptomatic therapy in NPC1 [37]. Miglustat is a small molecule that inhibits glycosylceramid synthase, one of the key components of the glycosphingolipid biosynthesis, therefore reducing intracellular lipid storage [38]. Long-term therapy with miglustat has been shown to increase lifespan and stabilize neurological functions. Additionally, miglustat has been ascribed activity against oxidative stress [39]. However, limitations consist in mainly gastrointestinal side effects such as diarrhea, weight decrease, and flatulence, but also tremor [40]. A further promising drug, 2-hydroxypropyl-β-cyclodextrin (HPβCD)—a cyclic oligosaccharide—is used as an enabling excipient in pharmaceutical formulations, as well as a cholesterol modifier in vivo. Therapy results in delayed onset of neurological symptoms with increased lifespan [37][38][41][37,38,41]. Matsuo et al. [42] reported in a clinical trial that HPβCD was effective in NPC1 patients, suggesting that HPβCD is a promising drug candidate in NPC1 disease. HPβCD overcomes the transport defect leading to excretion of accumulated cholesterol as bile acid, as shown in Npc1−/− mice [43]. It has been suggested that cholesterol efflux is mediated by the ATP binding cassette subfamily G member 1 (ABCG1), which promotes biliary excretion of sterols, ameliorating liver function [43][44][43,44]. Unfortunately, HPβCD administration also has side effects, particularly on the survival of outer hair cells, leading to hearing loss. This major side effect occurs in a dose- and duration-dependent manner [45][46][45,46]. What is more, in an open-label, dose-escalation phase 1–2a study, promising effects of HPβCD were recorded [47]; however, preliminary results of a current multinational phase 2b/3 clinical study involving about 50 patients treated with 200 mg/kg intrathecally applied HPβCD every 2 weeks indicate doubts that HPβCD achieves benefits when compared to a placebo [48][49][50][48,49,50].
Another promising therapy, so far applied only in animal models, consists of a combination of miglustat, the neurosteroid allopregnanolone, and HPβCD [13][51][52][53][13,51,52,53], resulting in further prevention of cerebellar Purkinje cell loss, improved motor function, reduced intracellular lipid storage, and prolonged life span in Npc1−/− mice.
Another therapeutic approach showed that the activity of the liver X receptor β (LXR β) can regulate the cholesterol flux from the brain, which leads to a reduction of neuroinflammation and slows therefore the neurodegeneration process. However, these positive effects result only in a modest lifespan prolongation [54][55][54,55]. Nevertheless, LXR β activation by treatment with an LXR agonist (T1317) can be useful in combination, e. g., with HPβCD.
In the absence of a causal treatment, there is still, a need to identify novel treatment strategies. Currently, histone deacetylase inhibitors (HDACi) are a focus of interest, due to the findings that they can reduce cholesterol accumulation in LE/LY [55][56][57][58][55,56,57,58]. These enzymes mediate posttranslational deacetylation of many types of proteins, e.g., histones, transcription factors, and chaperones [59]. In spite of its interaction with many different proteins and signaling pathways, it has been shown that HDACi increases expression of the low-activity mutant NPC1 protein [56][57][56,57], at least in vitro.
A further treatment option is FTY720 (fingolimod), a sphingosin analog. This drug is already approved for human use to treat multiple sclerosis [60]. FTY720 can enter the cell nucleus, where it is phosphorylated by sphingosine kinase 2 (SphK2). This active form is an inhibitor of class I histone deacetylases. The advantage of this drug over available HDACi is to regulate the expression of only a limited number of genes, which are restricted to cholesterol and sphingolipid metabolism, compared with the large number (thousands of genes [61]), which are activated by HDACi [60].
Another treatment approach is the application of arimoclomol, a coinducer of heat shock protein 70 (HSP 70) that improves the binding of several sphingolipid-degrading enzymes to their essential cofactor bis(monoacyl)glycerophosphate in vitro [62][63][62,63]. Beneficial effects for NPC patients have also been observed with drugs such as ursodeoxycholic acid [64][65][64,65] and acetyl-DL-leucine [66].
Moreover, an increased level of functional NPC1 can be achieved using gene therapy [67]. In some studies, it has been shown that the adeno-associated virus (AAV) 9 vector may successfully transfer the NPC1 gene into the CNS of Npc1−/− mice [67][68][69][67,68,69]. Systemic delivery of a functional NPC1 gene into Npc1−/− mice significantly extends the lifespan, ameliorates neurodegeneration, and improves behavioral abnormalities [67][68][69][67,68,69].