Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Giuliana Guggino and Version 2 by Catherine Yang.
Primary Sjogren syndrome (pSS) is a complex autoimmune disease mainly affecting salivary and lacrimal glands. Several factors contribute to pSS pathogenesis; in particular, innate immunity seems to play a key role in disease etiology. Invariant natural killer (NK) T cells (iNKT) are a T-cell subset able to recognize glycolipid antigens.
- Sjogren syndrome
- innate immunity
1. Primary Sjogren Syndrome
Primary Sjogren’s syndrome (pSS) is an autoimmune disease characterized by chronic inflammation of exocrine glands. In particular, salivary and lacrimal glands are the main target in pSS disease, characterized by a progressive lymphocytic infiltration causing the hallmarks symptoms of pSS: xerophthalmia and xerostomia [1].
The extraglandular manifestations (arthritis/arthralgias, vasculitis, and neuropathy affecting mainly peripheral nerves) can even occur in pSS underlining the systemic involvement of the disease [2].
Moreover, patients suffering from pSS are at high risk of developing B-cell non-Hodgkin’s lymphoma as a severe complication of the disease. It is estimated that approximately 5% of patients can develop non-Hodgkin lymphoma, and the risk is considered 7- to 19-fold higher when pSS patients are compared to the general population [3]. The most frequently identified type of lymphoma affects MALT (mucosal-associated lymphoid tissue) in salivary glands; however, more aggressive subtypes of lymphomas as large diffuse B-cell variants have been described [4]. Tobon et al. showed that Fms-like tyrosine kinase 3 ligand (Flt-3L) might be associated with lymphoma in pSS, suggesting its role as a biomarker to identify patients at high risk to develop proliferative disease [5]. Interestingly, in studies focused on salivary gland histology, another possible predictor of lymphoma development was identified. Low miRNA200b-5p levels were identified in pre-lymphoma pSS patients several years before the onset of hematological disease [6]. The prevalence of pSS was about 0.5% with a mean onset age in the 4th–5th decade, even if a later onset between the 6th–7th decade was quite common. Like other autoimmune diseases, pSS affects prevalently women with a female to male ratio of 9:1 [7].
pSS pathogenesis appears to be multifactorial: immune system dysregulation, genetic, epigenetic, environmental, infectious, and hormonal factors could play a role in the complex etiology of the disease. Several chemokines and cytokines account for both an inflammatory (CXCL13 CXCL10, CXCL8, CCL2, IL-10, and IL-6) and a pro-fibrotic (CXCL14, CCL28, tumor necrosis factor-related apoptosis-inducing ligand, and TGF β) effect. This intricate milieu finally triggers an abnormal immune response involving both innate and adaptive immune cells, leading to an initial process of epithelitis. In target tissues, the presence of ectopic lymphoid structures can be easily identified with evidence of B-lymphocyte expansion and the production of autoantibodies. This confirms the key role of adaptive autoimmunity in pSS at least in advanced phases of the disease [8].
To date, the search for reliable biomarkers able to ameliorate the diagnostic algorithm and the prognostic stratification of pSS patients is a major topic in pSS and remains an unmet need.
Autoantibodies, anti-Ro e anti-La, are classically considered hallmarks of pSS. However, new autoantibodies, known as tissue-specific antibodies (TSAs), are emerging as possible new biomarkers. TSAs include anti-salivary protein 1 (SP1) and anti-carbonic anhydrase 6 (CA6) that are associated with early stages of pSS and worse ocular manifestations, and anti-calponin antibodies are more related to peripheral neuropathy. Larger studies are needed to confirm the utility of these antibodies in routine clinical practice [9][10][9,10]. Considering pathogenesis, interferon (IFN) type I seems to be a central actor in pSS. It induces a wide range of genes involved in pro-inflammatory immune response and correlates with a more severe disease course [11]. This overexpression of IFN-related genes was described in both blood and salivary glands of pSS patients, characterizing the so-called IFN-signature in this specific rheumatic disease. Thus, IFN type I and its surrogates, as myxovirus-resistance protein A or the identification of IFN-induced genes, may represent possible biomarkers of pSS [12][13][12,13]. Among chemokines, CXCL13, which can be detected in peripheral blood as well as in salivary gland tissue, was demonstrated to correlate with disease activity and lymphoma development. This observation makes CXCL13 an attractive biomarker to monitor pSS progression [14].
Screening the non-invasive biomarkers from the saliva and tears has a significant potential too. These fluids represent a crucial source of valuable biomarkers as LACTO or LIPOC-1 tear proteins or S100A8/A9, a molecule found in the saliva of pSS that correlates with a higher risk of developing lymphoma [15][16][15,16].
Even the imaging techniques are considered future possible biomarkers of disease. Salivary glands ultrasonography stands out as the most promising field, and great research efforts are currently ongoing to define a clear possible role of this technique in pSS diagnosis, follow-up, and response to treatment. Particularly, elastosonography, which has the ability to measure the degree of glandular elasticity, seems to be a reliable tool to identify pSS patients with high sensitivity, specificity, and negative predictive value [17].
2. iNKT in Sjogren Syndrome
A rapid communication published in 2001 evidenced a decreased number of iNKT among patients suffering from autoimmune diseases, including pSS. A possible relation between low iNKT percentage and autoreactive tissue damage was addressed. The number of iNKT cells was proven to be significantly reduced in pSS, as well as in RA and SLE, while it was normal in diseases that commonly do not imply structural damages as myasthenia gravis and Graves disease. As a limit, only circulating iNKT was evaluated without analyzing tissue specimens of salivary glands [18][44].
Nevertheless, iNKT was also observed in pSS salivary gland tissues using CD3+CD16+ and CD56+ as markers to identify these cells. However, according to literature, these markers are not sufficient to identify iNKT [19][20][64,65]. After several years, another paper aimed to assess the frequency and function of iNKT in peripheral blood and salivary glands tissue from pSS patients, trying to identify iNKT cells using a tetramer construct. In detail, α-GalCer associated with a specific probe (CD1d/α-GalCer) was used to interact and bind specifically the invariant TCR of iNKT [19][64]. At the tissue level, iNKT was undetectable, and a high number of autoreactive B cells were identified. So, the different evaluation could be due to a technical difference in iNKT detection.
On the other hand, iNKT was expanded in peripheral blood samples from pSS patients. These cells stimulated with α-GalCer produced both IL-17 and IFN-γ. Interestingly, low levels of chemokine receptors as CXCR3, CCR6, and CCR5 were detected on the iNKT cells’ surface. This evidence may justify the impairment of activated iNKT to migrate towards inflammatory tissues. Specifically, in pSS, their absence in immune cells infiltrates in salivary glands can explain the lack of control on autoantibodies in situ production (Figure 1). The co-culture of B cells from pSS salivary glands with iNKT demonstrated that anti-SSA production was reduced in the presence of activated iNKT. This inhibitory activity on autoreactive B cells correlated with the number of iNKT in cultures [19][64]. The scientific background was mainly derived from the previously described results obtained in SLE. In particular, a reduction or the absence of iNKT in this autoimmune rheumatic disease was related to an increase in autoantibodies production, confirming the protective role of iNKT as negative regulators of autoantibodies producing B cells [21][66]. Taking into account the ability of iNKT to perform their regulatory function on self-reactive B cells, safeguarding non-self-reactive B cells [22][28], a possible future role, as a valid therapeutic target for iNKT emerges.
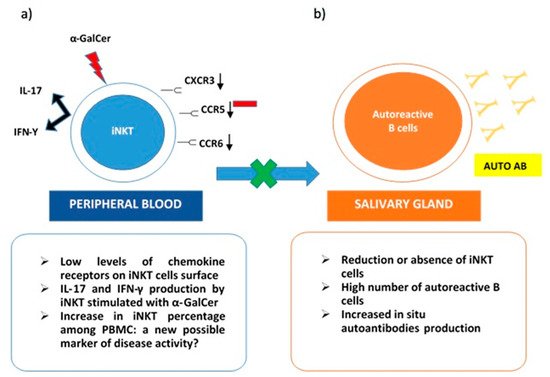
Figure 1. iNKT cells in Sjogren Syndrome. (a) in peripheral blood iNKT express low levels of chemokine receptors (CXCR3, CCR6, CCR5) on their surface. This could explain their reduced presence in salivary glands due to impaired migration. iNKT from pSS peripheral blood samples stimulated with a-GalCer produce both IL-17 and IFN-γ; (b) in the salivary gland, reduction or absence of iNKT cells determines the loss of their role as negative regulators on autoreactive B cells and the consequent in situ production of autoantibodies.
However, a deeper characterization of iNKT cytokine/chemokine profile and their surface receptors assessment, as well as of their migratory pattern, is required to identify a potential therapeutic option based on iNKT.
In addition to this, another strength point is the increased percentage of iNKT among PBMC in pSS. The number of NKT cells was found higher in pSS patients who had extraglandular manifestation, while the amount of NKT cells in patients without systemic involvement and controls was similar. Such a result could be due to an extreme attempt to counter-regulate an important ongoing inflammatory process. Furthermore, a significant increase in T regulatory type 1 (Tr1) cells and CD4+ CD25+ Treg cells was observed in patients with pSS with extraglandular manifestations compared to patients without these features. This data would seem to further confirm the presence of a possible compensatory mechanism, especially in the more severe forms of pSS with systemic inflammatory status [23][67].
It is well known that in pSS, there is a lack of markers of disease activity, and the evaluation of iNKT in peripheral blood could become an effective marker to assess it [19][64].
On the other hand, other studies investigated the presence of iNKT only among PBMC, obtaining, once again, the conflicting results. Several studies found a decrease in the number of iNKT in peripheral blood samples of pSS patients, while other researchers determined that the same cells were increased in pSS. In particular, Sudzuis et al. showed a significant reduction in the absolute NKT cell count in peripheral blood of patients with seropositive pSS, possibly ascribed to genetically determined lymphopenia. Furthermore, it has been hypothesized that this reduction could be due to migration in inflammatory sites or to apoptosis [24][68].
Moreover, two different groups of patients: responder and non-responder to α-GalCer in vitro stimulation were studied. In responder patients, a possible cause of iNKT global reduction could be an insufficient amount of natural ligands on APC. On the other hand, possible causes of iNKT unresponsiveness could be related to a specific dysfunction of iNKT or their overstimulation in a chronic inflammatory autoimmune environment. In this case, their prolonged activation could lead to a selective decrease in their number [25][24].
In addition to this, a paper published in 2017 by Davies et al. tried to better characterize peripheral leukocyte population of patients with pSS. Patients were further stratified according to the presence or absence of autoantibodies SSA/SSB and extraglandular manifestations. Researchers evidenced that the pSS patients, in particular, if seropositive and affected by systemic symptoms, presented a significant decrease in lymphocyte subpopulation when compared to controls. Conversely, the same patients appeared to have a higher number of granulocyte and monocyte subpopulations. Taking into account iNKT cells, an increase in this specific subgroup was found out in patients affected by a milder disease without important extraglandular involvement [26][69]. This interesting result, once again, confirms a possible protective role of NKT cells in the disease process leading to pSS.
In conclusion, it seems important to underline that different clusters of disease exist, and complete characterization of patients, stratified considering cellular and cytokine/chemokine profile, is mandatory to better understand disease phenotypes and subsequently to approach a specific targeted therapy [27][70].